2018-10-11 21:06:25 [runtime] (update) ID.normal2.SC_RNA_COUNTER_CS.SC_RNA_COUNTER._BASIC_SC_RNA_COUNTER.CHUNK_READS.fork0 chunks_running
2018-10-11 21:12:26 [runtime] (update) ID.normal2.SC_RNA_COUNTER_CS.SC_RNA_COUNTER._BASIC_SC_RNA_COUNTER.CHUNK_READS.fork0 chunks_running
2018-10-11 21:15:08 [runtime] (failed) ID.normal2.SC_RNA_COUNTER_CS.SC_RNA_COUNTER._BASIC_SC_RNA_COUNTER.CHUNK_READS
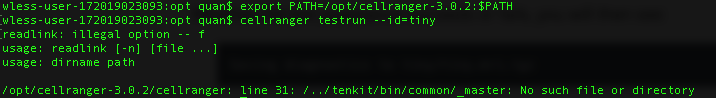
[error] Pipestance failed. Error log at:
normal2/SC_RNA_COUNTER_CS/SC_RNA_COUNTER/_BASIC_SC_RNA_COUNTER/CHUNK_READS/fork0/chnk0-u55bdbf4968/_errors
Log message:
Traceback (most recent call last):
File "/test/software/cellranger-2.1.1/martian-cs/2.3.2/adapters/python/martian_shell.py", line 529, in _main
stage.main()
File "/test/software/cellranger-2.1.1/martian-cs/2.3.2/adapters/python/martian_shell.py", line 495, in main
self._run(lambda: self._module.main(args, outs))
File "/test/software/cellranger-2.1.1/martian-cs/2.3.2/adapters/python/martian_shell.py", line 464, in _run
cmd()
File "/test/software/cellranger-2.1.1/martian-cs/2.3.2/adapters/python/martian_shell.py", line 495, in <lambda>
self._run(lambda: self._module.main(args, outs))
File "/test/software/cellranger-2.1.1/cellranger-cs/2.1.1/mro/stages/common/chunk_reads/__init__.py", line 53, in main
tk_subproc.check_call(chunk_reads_args)
File "/test/software/cellranger-2.1.1/cellranger-cs/2.1.1/tenkit/lib/python/tenkit/log_subprocess.py", line 37, in check_call
return subprocess.check_call(*args, **kwargs)
File "/test/software/cellranger-2.1.1/miniconda-cr-cs/4.3.21-miniconda-cr-cs-c9/lib/python2.7/subprocess.py", line 186, in check_call
raise CalledProcessError(retcode, cmd)
CalledProcessError: Command '['chunk_reads', '--reads-per-fastq', '5000000', '/test/XX/RNAseq/Batch2_normal/normal2/SC_RNA_COUNTER_CS/SC_RNA_COUNTER/_BASIC_SC_RNA_COUNTER/CHUNK_READS/fork0/chnk0-u55bdbf4968/files/', 'fastq_chunk', '--martian-args', 'chunk_args.json', '--compress', 'lz4']' returned non-zero exit status 1



































































































































{kind=link}