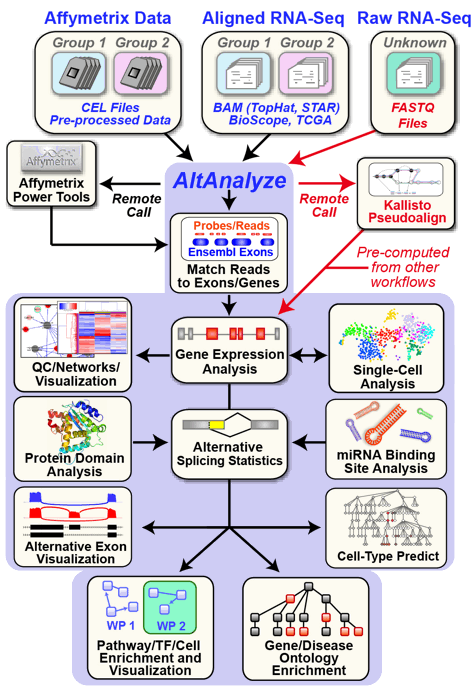
An automated cross-platform workflow for RNA-Seq gene, splicing and pathway analysis
AltAnalyze is an extremely user-friendly and open-source analysis tool that can be used for a broad range of genomics analyses. These analyses include the direct processing of raw single-cell and bulk RNASeq or microarray data files, advanced methods for single-cell population discovery and dataset comparison, differential expression analyses, analysis of alternative splicing/promoter/polyadenylation and advanced isoform function prediction analysis (protein, domain and microRNA targeting). Multiple advanced visualization tools and à la carte analysis methods are supported in AltAnalyze (e.g., network, pathway, splicing graph). AltAnalyze is compatible with various data inputs for RNASeq data (FASTQ, BAM, BED), microarray platforms (Gene 1.0, Exon 1.0, junction and 3' arrays) for automated gene expression and splicing analysis. This software requires no advanced knowledge of bioinformatics programs or scripting or advanced computer hardware. User friendly videos, online tutorials and blog posts are also available.
If installed from PyPI (pip install AltAnalyze), the below dependencies should be included in the installed package. When running from source code you will need to install the following libraries.
- Required: Python 2.7, numpy, scipy, matplotlib, sklearn (scikit-learn)
- Recommended: umap-learn, nimfa, numba, python-louvain, annoy, networkx, R 3+, fastcluster, pillow, pysam, requests, python-igraph, cairo
AltAnalyze documentation, stand-alone archives are provided at sourceforge as well as at github. For questions not addressed here, please contact us.
News Update 5/17/20