bxlab / galaxy-hackathon Goto Github PK
View Code? Open in Web Editor NEWThis project forked from galaxyproject/galaxy
Data intensive science for everyone.
Home Page: https://galaxyproject.org/
License: Other
This project forked from galaxyproject/galaxy
Data intensive science for everyone.
Home Page: https://galaxyproject.org/
License: Other
























































Galaxy can upload workflows to myexperiment.org but it would be also nice to upload workflows directly to the Galaxy ToolShed. For the sake of simplicity we should assume a TS account for the hackathon.
Things you can learn:
Skills:
As discussed with @frederikcoppens it would be nice to upload compressed FASTQ-files and to handle them properly in tools.
One way to do this is to enhance the already existing FASTQ datatype with a new metadata element that indicates if it compressed or not. Tools should then be able to recognize this metadata element. GSNAP is one of the tools which would benefit from it.
You can learn about:
Skills:
Working on fork of planemo (https://github.com/nturaga/planemo/tree/add_bioc_tool_init)
planemo bioc_tool_init --rscipt#Tool_development #Python #UCSC_trackHub #Visualization #Tracks #Cheetah
Hey!
Another project we could work on is...TrackHub UCSC Visualization through Galaxy:
Track hubs are web-accessible directories of genomic data that can be viewed on the UCSC Genome Browser.
Track hubs can be displayed on genomes that UCSC directly supports, or on your own sequence. Hubs are a useful tool for visualizing a large number of genome-wide data sets. For example, a project that has produced several wiggle plots of data can use the hub utility to organize the tracks into composite and super-tracks, making it possible to show the data for a large collection of tissues and experimental conditions in a visually elegant way, similar to how the ENCODE native data tracks are displayed in the browser.
Here are a few screenshots about what we have available:


The project is already well advanced, and you can see the repository here: https://github.com/goeckslab/hub-archive-creator
The TODO is here, but new ideas are more than welcome :)
You can also already play with it on this instance, under the G-OnRamp category.
Will be glad to work with you, guys!
Hey!
So with @bgruening and @erasche, we are implementing a chat inside Galaxy for users to discuss all together, and have private rooms. If you are interested in such a way of people interacting right inside Galaxy, please come to talk with all three of us and take a look at the PR we made into Galaxy branch:
The project contains Python / Javascript / Galaxy communication so everyone could contribute :)
Contributors: @jennaj @griffinp @kpoterlo @yvanlebras @BoughAida @ssander5 @devikaatgit @cschu @tnabtaf @kmurat1
This issue is dedicated to Training Data hackathon group. The idea is to gather sample data who can be used as example, tutorial, .... on Galaxy instances.
Please, don't hesitate to create a comment and add data links and description ;)
Example:
-parents
female http://546969.197.189.163/datasets/bbbfa414ae315caf/display/
male http://546969.197.189.163/datasets/4467809fea030689/display/
-progeny
progeny 1 http://546969.197.189.163/datasets/ddf83cf807e6e774/display/
progeny 2 http://546969.197.189.163/datasets/30bf7a4ced2335cc/display/
....
barcode http://546969.197.189.163/datasets/6df0b7b066ddc4c9/display/
population map http://546969.197.189.163/datasets/d796ca8e1687a54b/display/
reference genome http://546969.197.189.163/datasets/06cf32e9aa8aad75/display/
FastQ file http://546969.197.189.163/datasets/34c3e3c01e1a37f4/display/
If data are not reachable through the web (personal data on your laptop, ...) , the best way is to upload the data on a https://usegalaxy.org/ Galaxy history
The idea can be to meet after having gathering data and discuss about which one are good / duplicate / too big before proposing actions like, data directly shareable, need to be reduced, ....
See common-workflow-language/common-workflow-language#104
Tasks: produce Galaxy workflow following the instructions at https://github.com/nekrut/galaxy/wiki/Reference-based-RNA-seq
Repeat the above using the latest draft-4 syntax of CWL
We have implemented a Neo4j GIE based on a modified version of the Neo4j:2.3.3 image which takes a neostore datatype generated by this tool allowing users to explore a Neo4j Graph database. We have a demo galaxy instance with the IE here.
Goal:
Design representative testing/tutorial/training data to be published as Shared Data Libraries.
Objectives:
Discussion points:
Related
Add an output for splice junctions
2016 GCC Data Hackathon Hub
The 2nd annual Data Wrangling Hackathon (“Datathon”) starts on Saturday, June 25 and continues on Sunday, June 26. The associated Coding Hackathon #1 overlaps with these dates. The working rooms of both hacks will be in close proximity and the two Hackathons will share meals to encourage and facilitate collaboration.
https://gcc2016.iu.edu/hacks/index.php
https://gcc2016.iu.edu/hacks/hacks-data.php
Goals
Datathon Focus
Code Hack Focus Collaboration
Those participating in the GCC 2016 Datathon will have the opportunity to contribute to Galaxy in the following areas:
Aside from the aforementioned areas, all forms of contribution are welcome! Everyone is strongly encouraged to post suggestion/opinion/criticism below.
We will be using Github to organize this year (not Trello). This is how it works:
See all soon! Cheers from Christian (@cschu), Frederik (@frederikcoppens), and Jennifer (@jennaj)
For prior comments, please see: galaxyproject#2520
E Rasche's (@erasche) JBrowse tool is pretty awesome.. The idea here is to add some functionality to the tool.
Should be a small job.
As part of the #26 we needed to create a neostore composite datatype. This is similar to blastdb composite datatype. This datatype is for a neo4j database which is generated by this tool https://github.com/SANBI-SA/tools-sanbi-uwc/tree/master/tools/build_ctb_gene currently in the testtoolshed.
The datatype is here: https://github.com/SANBI-SA/tools-sanbi-uwc/tree/master/datatypes/neo4j_datatypes
Comment on this issue and I'll add your user to the hackathon team so you'll be able to create branches here and commit.
Also, please go ahead and make planning issues for any projects you'd like to work on!
A colleague has written a python script that uses the data library api to add data libraries of microbial references to Galaxy by genus and/or species from Refseq.
@bwlang and me have decided to check if we can
The API endpoint already works, although strikingly a GET causes the dependency to be resolved and installed.
r = requests.get(url+"/api/dependency_resolvers/0/requirements", params={"key": your_api_key, "name": "bowtie2==2.2.8"})
One difficulty here is that the tool shed takes the dependency information (or requirements) from the tool_dependencies.xml file (which I guess will be phased out in new tools at some point).
So if we want to do that nice "Install dependencies / requirements " the toolshed should probably return the requirements from the tool xml.
And indeed, the toolsshed does already provide that information through the API
r = requests.get(ts_url+"/api/repositories/get_repository_revision_install_info", {"name":"msp_sr_readmap_and_size_histograms", "owner":"drosofff", "changeset_revision":"d6b93af0da55"})
Make changes to the galaxy to allow access from IGV.js
retrieve external dataset url for bed,gff,bam,vcf from api
Python 2.7 will not be maintained past 2020.
Add support for Python >= 3.3 while maintaining support for Python 2.6 and 2.7.
xref.: https://trello.com/c/dZcCVf9I/2702-migrate-to-python-3
The list of files already fixed is in .ci/py3_sources.txt
Useful tools and documentation:
https://docs.python.org/3/howto/pyporting.html
https://docs.python.org/2/library/2to3.html
https://python-modernize.readthedocs.org/
https://pythonhosted.org/six/
http://python3porting.com/preparing.html
Requirements which need to be ported or dropped:
xref: galaxyproject#1715
I'm working on (https://github.com/dannon/galaxy/tree/quota_display) a better way for users to view and delete the data they have. I have a work in progress branch that uses d3 to generate a treemap, and plotly to display it. It looks like this:

Would love help. It's primarily javascript w/ backbone, though we might have to add functionality to the API in Galaxy, which would of course be work in python.
Primary targets are better hover information, the ability to select histories from the view, and general usability improvements prior to linking it into Galaxy.
Last year we started Galaxy Scientists as a forum for power uses to discuss UX improvements.
This did not really take of using Slack.
Is it worth to try to revive this? e.g. through a galaxy/galaxy_scientists github repo and discuss there?
We have Galaxy instance running in our laboratory for more than 4 years. Recently, we ran out of disk space (100TB) and then we realised the FASTQ files occupied more than 60% of our disk space. I would like whether there is any solution to compress the existing FASTQ datasets both in histories and libraries of the galaxy.
Provide a way to label history items, and have the labels propagate downstream to derivative history items.
In other words: Show sample names on history items, which is much more useful than sticking "on data 11" onto the title.
UI:
Show each label in a box off to the side of the title.
Implementation:
Store in a new database table
There should be a data manager for genome annotation files to populate the .loc file (e.g. to use for featurecounts).
@davebx ?
Associated Coding Hack Tasks
Recently we integrated shared form building procedures into Charts, this allows us to use color pickers and sliders to configure visualization. This hackathon project intends to demonstrate this by adding color pickers to specify series colors.
POC of steps needed to
To Do
make an interactive tour for this too?
Working on this here .. will try to publish to the toolshed as needed:
Trinotate is a transcriptome annotation and quantification pipeline that stores the results in a sqlite database. The database contains transcript to gene mapping, protein predictions, blast matches for nucleotides and proteins against multiple databases, several prediction software tools results (signalp etc) and differential expression results.
Trinotate web provides an interface to the results but requires a web server and sqlite. The interface doesn’t support filtering or pagination but only keyword search in blobs of data combining all results for a given transcript.
I have created a galaxy visualisation for Trinotate sqlite db. Currently it can display the contents of the db with filtering and pagination. The source code is in https://github.com/Eduardo-Alves/trinotateviz.git The test folder contains a sample trinotate sqlite database provided with Trinotate Web.
My plan is to add the ability to compare multiple transcriptomes and use galaxy trackster browser to see how different transcriptomes align to uniref proteins or a related species. I have several questions on how to implement this:
1-The grid was implemented using angular.js ui-grid because of the support for expandable grids. Is this a good choice? Any better alternatives?
2-Server side filtering and pagination was implemented in the sql call to sqlite data provider. In order to keep the various sub grids in sync all results include gene_id subquery that joins all tables and filters. This is quite messy. Again is there any better alternative?
3-The first improvement I need is to be able to compare multiple transcriptomes. I don’t want to modify the database schema so to maintain compatibility with trinotate. I am planning to pre- append the gene names with transcriptome name and then filter in sql. Each assembly will have a sub-grid and filtering will be done across all transcriptome so that it will be possible to see for instance how many genes are differentially expressed in each assembly or how many of these have blast results for a given e-value threshold. Is the subgrid idea reasonable?
4-Next requirement is to be able to visualise transcripts from various transcriptomes against a single reference, which can be alignments of predicted proteins against uniref. This will allow researchers to see which transcriptomes best assemble their proteins of interest. The idea is to use trackster to display blast results of transcripts aligning to a given protein. I haven’t started looking into this yet so any ideas are welcome.
Any suggestions, bug fixes, pull requests are welcome. This is my first javascript project so please forgive in advance the bugs.

I'd like to implement an optional default naming convention that includes
*tool name* on *original input dataset*
@mvdbeek thinks this could be implemented with the element identifier field being talked about:
galaxyproject#2006
galaxyproject#2140
Perhaps as a next phase we could find a way to designate an input dataset or collection as a "naming source"
I'd like to find a way to make this work in the context of a workflow, but also in the context of ad-hoc analysis.
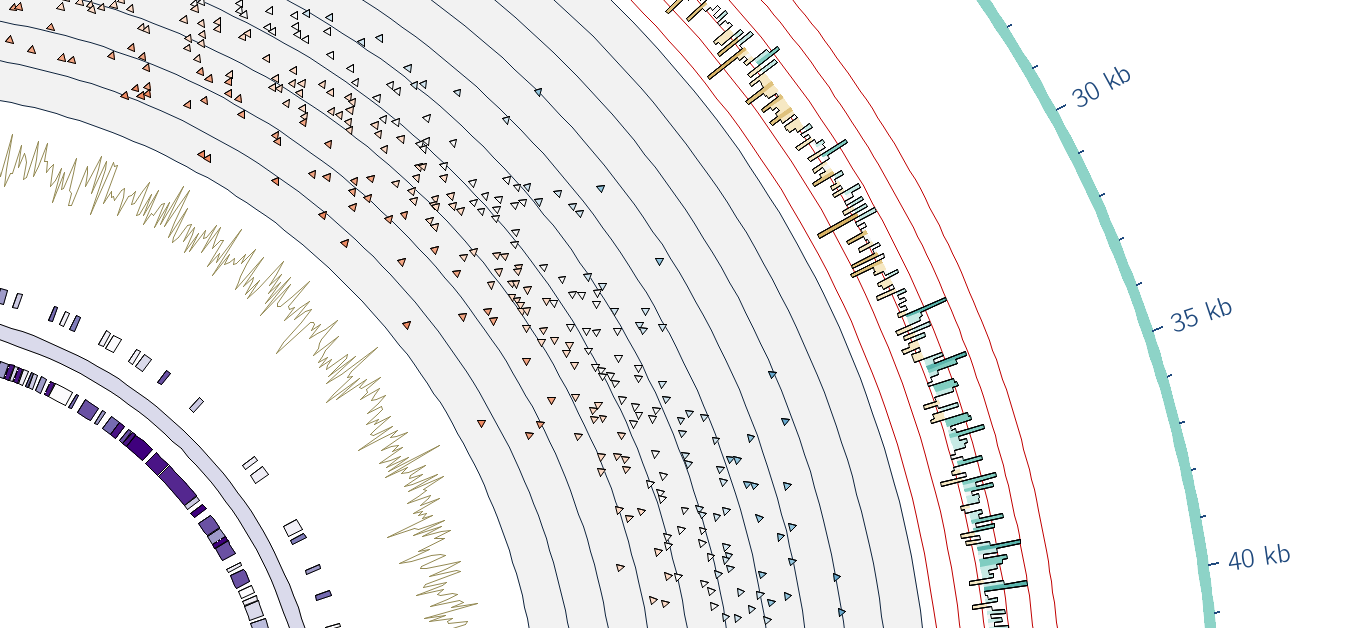
I've built a Circos tool. If you'd like to play around with it and give feedback, just say hi and I've got a copy installed.
http://circos.ca
https://github.com/TAMU-CPT/galaxy-circos-tool
Stringtie was updated and now includes a merge option, providing an alternative to Cuffmerge/Cuffcompare.
I think @davebx has started wrapping the new version.
When installing a workflow with dependencies, the tools are installed again while they were already available in the instance.
Can this be avoided?
I did not specify versions in the xml for dependencies (in case this would make a difference)
Adding more support for password strength, changes and handling; inactive account suspension, and adding account creation with tokens and email to bioblend.
It would be nice to support uploading file larger than 2GB. For example the http://resumablejs.com library can be used.
You can learn about:
Skills:
When installing a tool, you need to separately install the data managers to generate indices, ... Could this be added as a (default on) option upon tool install?
Uncompressed fastq is a huge waste of disk space. So, a proposal:
Finish up this galaxyproject/starforge#95 and then this: galaxyproject#2385
Package uWSGI in a standalone wheel (pyuwsgi). Make it the default for checked out Galaxy (already will be default for packaged Galaxy).
This would mean IEs will require uwsgi. We can enable features conditionally based on whether the uwsgi module is available.
Stretch goal: create some endpoint that uses websockets for testing.
Allow trackster to export the current view rendered as SVG rather than a bitmap.
Please add any tools that you're adding to conda below. Feel free to modify this comment to add things.
An all new CloudLaunch is under development - multiple pluggable applications (with versions), multiple clouds, list of public Galaxy instances (+eventual search across those), and other features you would like to add.
The backend is done in Django (https://github.com/galaxyproject/cloudlaunch/tree/dev), exposing a REST API, and the front-end is done in Angular2 (https://github.com/galaxyproject/cloudlaunch-ui).
Feel free to join and add your components or help polish the existing functionality.

(Subject ref: https://en.wikipedia.org/wiki/All_your_base_are_belong_to_us)
Name, Permissions, Directory structure, Annotation (Lib and datasets), Metadata (database, datatype) and all other attributes preserved.
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.