pcingola / snpsift Goto Github PK
View Code? Open in Web Editor NEWLicense: Other
License: Other






































Hi,
I want to extract '.' from float fields, however snpSift seem to auto-convert it to 0.0 anyway. That will cause confusion with 0 for other records. Tried '-e .', and it give the same result.
Is there a way to skip the auto-conversion?
Many thanks.
❯ SnpSift
SnpSift version 4.3t (build 2017-11-24 10:18), by Pablo Cingolani
❯ cat /tmp/a.vcf
##fileformat=VCFv4.2
##FORMAT=<ID=GT,Number=1,Type=String,Description="Genotype">
##INFO=<ID=AF_popmax,Number=.,Type=Float,Description="AF_popmax annotation provided by ANNOVAR">
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT
chr1 16964 . T C 50.7 . AF_popmax=. GT 0/1❯ SnpSift extractFields /tmp/a.vcf AF_popmax
AF_popmax
0.0According to the VCF spec. 4.2, missing requires genotypes fields have to comply with ploidy.
So '.' is incorrect and './.' should be used for diploid organisms.
When rewriting for ANTLR4, keep in mind the nested expression problem.
Dear,
I'm using SnpEff and SnpSift to annotate a vcf file containing biallelic and multiallelic variants. For multiallelic, variants i.e. C/A,T, SnpEff annotates each possible combination (C/A and C/T). On the other hand, SnpSift does only one annotation for this multiallelic variant, which means C/A,T . The SnpSift is considering only the C/A frequency when annotating C/A,T. Yet, there are only allele frequencies for C/A and C/T, but not for C/A,T in the database.
Rogério
Dear sir or madam,
I am sorry to interrupt you.
When I was using snpSift v3.6 to run a pipeline, I encountered an error.
Exception in thread "main" java.lang.RuntimeException: Missing 'chr' columns in dbNSFP file at ca.mcgill.mcb.pcingola.fileIterator.DbNsfpFileIterator.parseHeader(DbNsfpFileIterator.java:382) at ca.mcgill.mcb.pcingola.fileIterator.DbNsfpFileIterator.getFieldNames(DbNsfpFileIterator.java:143) at ca.mcgill.mcb.pcingola.snpSift.SnpSiftCmdDbNsfp.checkFieldsToAdd(SnpSiftCmdDbNsfp.java:158) at ca.mcgill.mcb.pcingola.snpSift.SnpSiftCmdDbNsfp.run(SnpSiftCmdDbNsfp.java:362) at ca.mcgill.mcb.pcingola.snpSift.SnpSift.run(SnpSift.java:233) at ca.mcgill.mcb.pcingola.snpSift.SnpSift.main(SnpSift.java:60)
The version of dbNSFP is 3.3a.
The first line of the dbNSFP file is
#Chr,Start,End,Ref,Alt,SIFT_score,SIFT_converted_rankscore,SIFT_pred,Polyphen2_HDIV_score,Polyphen2_HDIV_rankscore,Polyphen2_HDIV_pred,Polyphen2_HVAR_score,Polyphen2_HVAR_rankscore,Polyphen2_HVAR_pred,LRT_score,LRT_converted_rankscore,LRT_pred,MutationTaster_score,MutationTaster_converted_rankscore,MutationTaster_pred,MutationAssessor_score,MutationAssessor_score_rankscore,MutationAssessor_pred,FATHMM_score,FATHMM_converted_rankscore,FATHMM_pred,PROVEAN_score,PROVEAN_converted_rankscore,PROVEAN_pred,VEST3_score,VEST3_rankscore,MetaSVM_score,MetaSVM_rankscore,MetaSVM_pred,MetaLR_score,MetaLR_rankscore,MetaLR_pred,M-CAP_score,M-CAP_rankscore,M-CAP_pred,CADD_raw,CADD_raw_rankscore,CADD_phred,DANN_score,DANN_rankscore,fathmm-MKL_coding_score,fathmm-MKL_coding_rankscore,fathmm-MKL_coding_pred,Eigen_coding_or_noncoding,Eigen-raw,Eigen-PC-raw,GenoCanyon_score,GenoCanyon_score_rankscore,integrated_fitCons_score,integrated_fitCons_score_rankscore,integrated_confidence_value,GERP++_RS,GERP++_RS_rankscore,phyloP100way_vertebrate,phyloP100way_vertebrate_rankscore,phyloP20way_mammalian,phyloP20way_mammalian_rankscore,phastCons100way_vertebrate,phastCons100way_vertebrate_rankscore,phastCons20way_mammalian,phastCons20way_mammalian_rankscore,SiPhy_29way_logOdds,SiPhy_29way_logOdds_rankscore,Interpro_domain,GTEx_V6_gene,GTEx_V6_tissue
Hope you can give me some suggestions or help, thank you very much!!
Problem: Using '=' for annotations is a problem since there might be more than one annotated 'effect' (e.g. "5_prime_UTR_truncation&exon_loss_variant")
We need an operator that can express this in a sucint way:
cat my.vcf | snpsift filter " ANN[*].EFF has 'exon_loss_variant' "
In this case, the operator 'has' should try to match:
i) Full string equality
ii) Split using any of the predefined separators (e.g.: &, +, comma, colon, semicolon, pipe, parenthesis, brackets, etc.) and then check for equality on the separated strings.
$ SnpSift vcfCheck foo.vcf
Exception in thread "main" java.lang.NullPointerException
at org.snpsift.SnpSiftCmdVcfCheck.run(SnpSiftCmdVcfCheck.java:56)
at org.snpsift.SnpSift.run(SnpSift.java:586)
at org.snpsift.SnpSift.main(SnpSift.java:76)
This expression produces an error:
$ java -jar SnpSift.jar filter "ANN[*].IMPACT = 'HIGH' | ANN[*].IMPACT = 'MODERATE'" z.vcf
whereas this one works OK:
$ java -jar SnpSift.jar filter "(ANN[*].IMPACT = 'HIGH') | (ANN[*].IMPACT = 'MODERATE')" z.vcf
Apparently the 'or' operator and the 'equals' operator are being evaluated using an incorrect precedence.
SnpSift dbnsfp: Some of the columns in the database have multiple entries (e.g. SIFT_score). It looks like by default the INFO header line encodes NUMBER=A for the field.
##INFO=<ID=dbNSFP_SIFT_score,Number=A,Type=Float,Description="Field 'SIFT_score' from dbNSFP”>
Due to the variable number of entries for some of these scores in the dbNSFP data base, they fail validation. It can be fixed by changing the header to “Number=.” since the number of values is unknown.
I have a variant that has dashes in the name. This seems to break SnpSift.
$ java -jar downloaded-software/snpEff/SnpSift.jar
SnpSift version 5.0e (build 2021-03-09 06:01), by Pablo Cingolani
$ java -jar downloaded-software/snpEff/SnpSift.jar extractFields test-original.vcf GEN[Foo-f].GT
GEN[Foo-f].GT
Exception in thread "main" java.lang.RuntimeException: INFO field 'Foo' not found in VCF header
at org.snpsift.lang.expression.Field.getReturnType(Field.java:242)
at org.snpsift.lang.expression.Field.eval(Field.java:63)
at org.snpsift.lang.expression.ExpressionBinary.eval(ExpressionBinary.java:25)
at org.snpsift.lang.expression.FieldSub.evalIndex(FieldSub.java:35)
at org.snpsift.lang.expression.FieldSub.evalIndex(FieldSub.java:27)
at org.snpsift.lang.expression.FieldGenotype.evalGenotype(FieldGenotype.java:24)
at org.snpsift.lang.expression.FieldGenotype.getFieldString(FieldGenotype.java:49)
at org.snpsift.lang.expression.Field.eval(Field.java:76)
at org.snpsift.SnpSiftCmdExtractFields.evaluate(SnpSiftCmdExtractFields.java:47)
at org.snpsift.SnpSiftCmdExtractFields.run(SnpSiftCmdExtractFields.java:151)
at org.snpsift.SnpSiftCmdExtractFields.run(SnpSiftCmdExtractFields.java:122)
at org.snpsift.SnpSift.run(SnpSift.java:580)
at org.snpsift.SnpSift.main(SnpSift.java:76)
$ cat test-original.vcf | sed s/Foo-f/Foo_f/ | java -jar downloaded-software/snpEff/SnpSift.jar extractFields - CHROM POS GEN[Foo_f].GT
CHROM POS GEN[Foo_f].GT
01 6240445 0/0
01 6240565 0/0
01 6240588 0/1
The VCF 4.2 (Section 1.3) header line is as follows:
#CHROM POS ID REF ALT QUAL FILTER INFO FORMAT Foo-f Foo-i Foo-p
It is unclear to me what the VCF 4.2 specification means by 'String' but it seems reasonable that a genotype name could contain a dash or underscore.
User request: Add LRG annotations.
What is the correct way to read this output generated by SNPEff?
**
**
Specifically I'm dealing with an HIGH impact frame shift of a gene of interest.
Thank you
SC
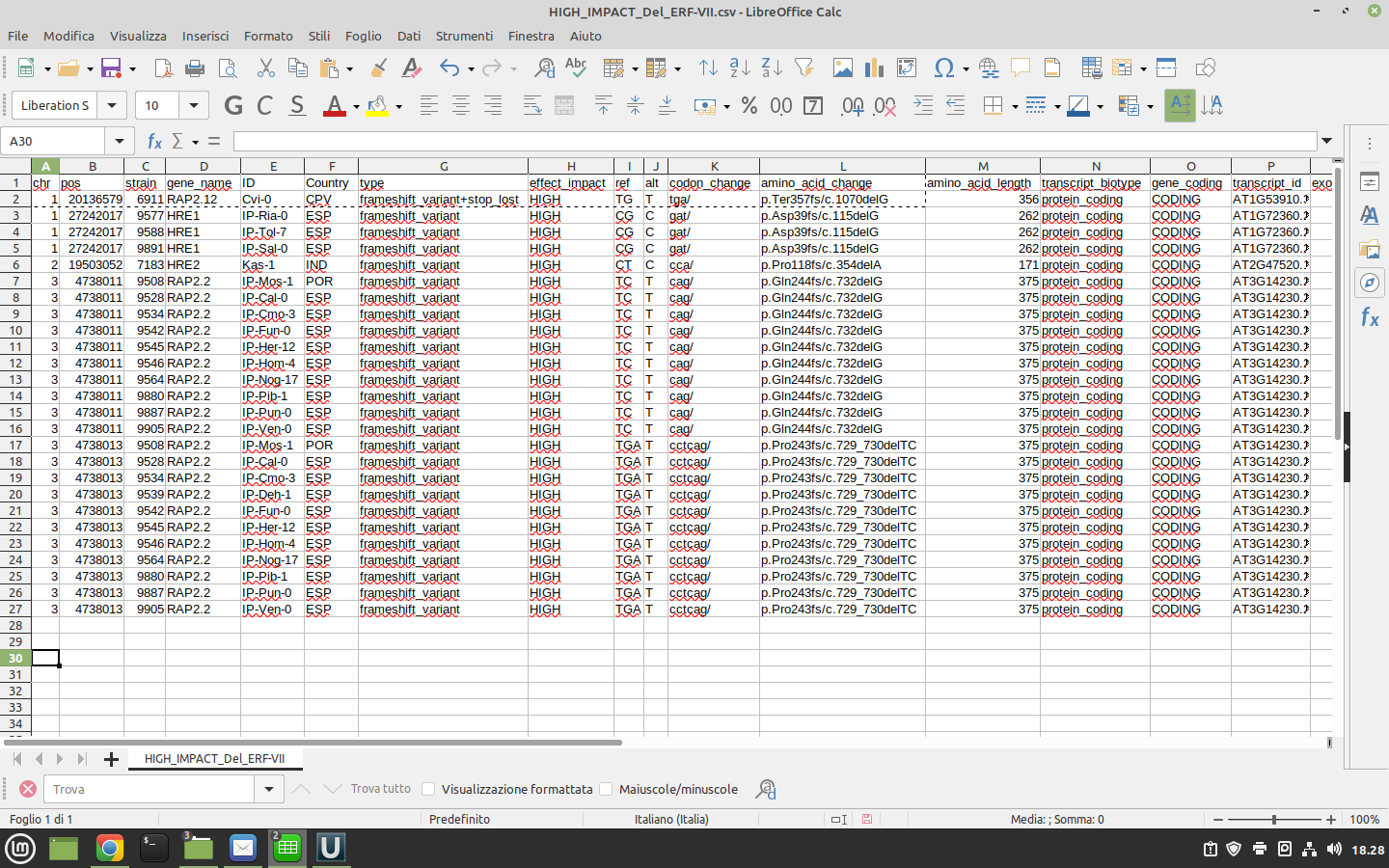
Hi,
I am trying to annotate some VCF files with some COSMIC data (latest realeve V91) but the INFO filed for the COSMIC vcf has "=" in it and snpsift throws this error:
java.lang.RuntimeException: No white-space, semi-colons, or equals-signs are permitted in INFO field values. Name:"AA" Value:"p.W263R,p.W344R,p.W235R,p.W343R,p.L211P,p.W218R,p.L116P,p.P288=,p.W281R"
I am on version 4.3T
SnpSift version 4.3t (build 2017-11-24 10:18), by Pablo Cingolani
Here is the header of the COSMIC vcf:
ryan@ubuntu:~/NGS/$ head -n 20 CosmicCodingMuts.vcf ##fileformat=VCFv4.1 ##source=COSMICv91 ##reference=GRCh38 ##fileDate=20200324 ##comment="Missing nucleotide details indicate ambiguity during curation process" ##comment="URL stub for ID field (use the whole COSV identifier)='https://cancer.sanger.ac.uk/cosmic/search?genome=38&q='" ##comment="REF and ALT sequences are both forward strand ##INFO=<ID=GENE,Number=1,Type=String,Description="Gene name"> ##INFO=<ID=STRAND,Number=1,Type=String,Description="Gene strand"> ##INFO=<ID=GENOMIC_ID,Number=1,Type=String,Description="Genomic Mutation ID"> ##INFO=<ID=LEGACY_ID,Number=1,Type=String,Description="Legacy Mutation ID"> ##INFO=<ID=CDS,Number=1,Type=String,Description="CDS annotation"> ##INFO=<ID=AA,Number=1,Type=String,Description="Peptide annotation"> ##INFO=<ID=HGVSC,Number=1,Type=String,Description="HGVS cds syntax"> ##INFO=<ID=HGVSP,Number=1,Type=String,Description="HGVS peptide syntax"> ##INFO=<ID=HGVSG,Number=1,Type=String,Description="HGVS genomic syntax"> ##INFO=<ID=CNT,Number=1,Type=Integer,Description="How many samples have this mutation"> ##INFO=<ID=SNP,Number=0,Type=Flag,Description="classified as SNP"> #CHROM POS ID REF ALT QUAL FILTER INFO 1 65797 COSV58737189 T C . . GENE=OR4F5_ENST00000641515;STRAND=+;LEGACY_ID=COSN23957695;SNP;CDS=c.9+224T>C;AA=p.?;HGVSC=ENST00000641515.2:c.9+224T>C;HGVSG=1:g.65797T>C;CNT=1
Thanks for any help you can provide!
Ryan
It's messing up all downstream analysis.
Instead of
$ snpsift extractfields s.vcf CHROM POS REF ALT LBS[*] | head
1 26996 A G 113 398 10 15 360 784 24 40
1 30860 G C 48 18 386 206 143 194 35 60
1 38232 A G 90 104 2 4 56 252 4 6
1 38907 C T 14 24 308 1004 25 25 282 1089
1 41218 T A 264 493 27 12 18 11 69 60
1 41256 C T 3 7 40 46 2 1 98 212
1 41981 A G 93 374 6 23 100 220 4 33
1 42577 C T 6 6 47 19 6 2 146 84
1 43586 C A 82 32 179 52 3 0 3 1
We should have an option to get:
1 26996 A G 113,398,10,15,360,784,24,40
1 30860 G C 48,18,386,206,143,194,35,60
1 38232 A G 90,104,2,4,56,252,4,6
1 38907 C T 14,24,308,1004,25,25,282,1089
1 41218 T A 264,493,27,12,18,11,69,60
1 41256 C T 3,7,40,46,2,1,98,212
1 41981 A G 93,374,6,23,100,220,4,33
1 42577 C T 6,6,47,19,6,2,146,84
1 43586 C A 82,32,179,52,3,0,3,1
Hi there - I have downloaded dbNSFPv4.1a from the website and am trying to create a database for snpSift to use. I followed the instructions on this page.
https://pcingola.github.io/SnpEff/ss_dbnsfp/
However, tabix is not able to index the file and throws error below
[get_intv] the following line cannot be parsed and skipped: Binary file (standard input) matches
[ti_index_core] the indexes overlap or are out of bounds
It appears like there are multiple records for one genomic position for different alternate alleles. I have tried to sort the file, check for empty lines and check for complete duplicate lines.
Please help.
Thank you,
Kuki
Hi Pablo,
Many thanks for all the updates to SnpSift. I'm trying to annotate using dbnsf 2.3, so I downloaded the gz file, created a tabix index and tried to run SnpSift. I keep running to the following error:
00:00:00.000 Annotating entries from: '/ngs/reference_data/genomes/Hsapiens/hg19/dbNSF/dbNSFP2.3/dbNSFP2.3.txt.gz'
WARNING: Field (column) 'SLR_test_statistic' does not have an approriate field descriptor.
Exception in thread "main" java.lang.ArrayIndexOutOfBoundsException: -1
at net.sf.samtools.tabix.TabixReader.query(TabixReader.java:360)
at net.sf.samtools.tabix.TabixReader.query(TabixReader.java:408)
at ca.mcgill.mcb.pcingola.fileIterator.MarkerFileIterator.seek(MarkerFileIterator.java:176)
at ca.mcgill.mcb.pcingola.snpSift.SnpSiftCmdDbNsfp.findDbEntry(SnpSiftCmdDbNsfp.java:239)
at ca.mcgill.mcb.pcingola.snpSift.SnpSiftCmdDbNsfp.annotate(SnpSiftCmdDbNsfp.java:101)
at ca.mcgill.mcb.pcingola.snpSift.SnpSiftCmdDbNsfp.run(SnpSiftCmdDbNsfp.java:347)
at ca.mcgill.mcb.pcingola.snpSift.SnpSift.run(SnpSift.java:233)
at ca.mcgill.mcb.pcingola.snpSift.SnpSift.main(SnpSift.java:60)
Any idea what this could be about? Here are the two first lines of the dbnsf file itself:
#chr pos(1-coor) ref alt aaref aaalt hg18_pos(1-coor) genename Uniprot_acc Uniprot_id Uniprot_aapos Interpro_domain cds_strand refcodon SLR_test_statistic codonpos fold-degenerate Ancestral_allele Ensembl_geneid Ensembl_transcriptid aapos aapos_SIFT aapos_FATHMM SIFT_score SIFT_score_converted SIFT_pred Polyphen2_HDIV_score Polyphen2_HDIV_pred Polyphen2_HVAR_score Polyphen2_HVAR_pred LRT_score LRT_score_converted LRT_pred MutationTaster_score MutationTaster_score_converted MutationTaster_pred MutationAssessor_score MutationAssessor_score_converted MutationAssessor_pred FATHMM_score FATHMM_score_converted FATHMM_pred RadialSVM_score RadialSVM_score_converted RadialSVM_pred LR_score LR_pred Reliability_index GERP++_NR GERP++_RS phyloP 29way_pi 29way_logOdds LRT_Omega UniSNP_ids 1000Gp1_AC 1000Gp1_AF 1000Gp1_AFR_AC 1000Gp1_AFR_AF 1000Gp1_EUR_AC 1000Gp1_EUR_AF 1000Gp1_AMR_AC 1000Gp1_AMR_AF 1000Gp1_ASN_AC 1000Gp1_ASN_AF ESP6500_AA_AF ESP6500_EA_AF
1 35138 T A X Y 25001 FAM138A . . . . - TAA . 32 T ENSG00000237613 ENST00000417324 86 . . . . . . . . .. . . 1 0.0 N . . . . . . . . . .. . 0.742 0.742 0.593000 0.0:0.0:0.0:1.0 3.8237 . . . . . . .. . . . . . .
Example from user (using dbSnp for bosTau):
WARNING: Reference in database file 'snp138.vcf' is 'NCA' and reference in input file is TCA' at 17:4302815
Here the database VCF file has REF='NCA' and input VCF file has REF='TCA' which causes a warning (and probably also causes missing annotation)
Problem
FILTER expression involving '*' or '?' does not give correct result if more than 1 variable used.
Example:
query Q1 => filter "(BAR[*] < 0.1)" test.vcf -> yields no result
query Q2 => filter "(BAR[*] < 0.1) & (FOO[*] >= 2)" test.vcf -> yields 3 result
Q2 is more strict than Q1 so its result should always be a subset of Q1's result.
Hi I discovered the above problem some time back and submitted a pull request
(#37)
Please let me know if this is the proper channel for submitting patches.
Thanks.
** Currently there's a merge conflict (minor issue due to test cases numbering). Once the PR is reviewed, I can incorporate feedback / rebase and resolve the conflicts.
When running SnpSift CaseControl, we observe variants that have high statistical significance, yet, when we inspect the variant in IGV, we often see that the samples we designated as our 'controls' that contain the variant outnumbers the number of samples we designated as 'cases' that have the variant.
Attached is 1) a table of variants generated from SnpSift extractFields where we pull in the CaseControl statistics among other fields we care about, 2) a shortened VCF (in .txt format in order to upload here) for the one variant example I am highlighting and 3) a screenshot of the "Variant Attributes" of this variant in IGV.
For this particular example, I want to highlight the variant in the variant table that is highlighted in yellow (gene CFH). When sorting ascending under the CC_DOM model, it has statistical significance to the negative 11th power. We have a total of 276 samples, 57 of which are "cases" and 191 which are "controls" (the other 28 are neutral/ignored). When we look in IGV, we see that there are 16 cases and 144 controls that show this heterozygous variant. I would like to understand the statistics calculations more for the models. One would assume that if more "control" samples have the variant than in the "case" samples, the variant would not have statistical significance to the negative 11th power so I was wondering if I could get a little more clarity so I can understand CaseControl more.
If there is anything else I can provide to help with the clarification, please let me know.
casecontrol_sorted_on_DOM.xlsx

Hello,
I was running concordance analysis via:
java -jar SnpSift.jar concordance -v old.vcf new.vcf > concordance.txt
I got my results in concordance_old_new.by_sample.txt
Can you please tell me where is the documentation or what is the meaning of the columns I got in the output of that file? and how can I actually compare results for samples from old.vcf and new.vcf, what are the most relevant parameters from this result?
[1] "sample" "MISSING_ENTRY_old.MISSING_ENTRY_new"
[3] "MISSING_ENTRY_old.MISSING_GT_new" "MISSING_ENTRY_old.REF"
[5] "MISSING_ENTRY_old.ALT_1" "MISSING_ENTRY_old.ALT_2"
[7] "MISSING_GT_old.MISSING_ENTRY_new" "MISSING_GT_old.MISSING_GT_new"
[9] "MISSING_GT_old.REF" "MISSING_GT_old.ALT_1"
[11] "MISSING_GT_old.ALT_2" "REF.MISSING_ENTRY_new"
[13] "REF.MISSING_GT_new" "REF.REF"
[15] "REF.ALT_1" "REF.ALT_2"
[17] "ALT_1.MISSING_ENTRY_new" "ALT_1.MISSING_GT_new"
[19] "ALT_1.REF" "ALT_1.ALT_1"
[21] "ALT_1.ALT_2" "ALT_2.MISSING_ENTRY_new"
[23] "ALT_2.MISSING_GT_new" "ALT_2.REF"
[25] "ALT_2.ALT_1" "ALT_2.ALT_2"
[27] "ERROR"
The '-name' prefix does not get appended to the vcf header info.
'Database VCF headers must be parsed and names changed accordingly (now headers are just copied form the 'database' VCF file).
Update old VCF "SnpSift annotate" to use new index.
From user:
I have a VCF file...When extracting the values of the format fields, the output is:
GEN[0].DP GEN[1].DP GEN[0].FDP GEN[1].FDP ...
Is it possible to have to automatically have the GEN[X] values be replaced with the actual name of the sample. So for example, have this:
NORMAL.DP TUMOR.DP NORMAL.FDP TUMOR.FDP ...
Just makes it easier to interpret the output.
I am trying to filter on a field with round parentheses in its name but I cannot figure out what character to use to escape them in the SnpSift filter query string.
dbscSNV from the authors of dbNSFP
Hello,
i followed the instructions on this page https://pcingola.github.io/SnpEff/ss_extractfields/ in order to use extractfields to separate annotations in separate columns.. I used this command:
$ cat my.ann1.vcf | /mnt/e/Bioinformatics/tools/snpeff/snpEff/scripts/vcfEffOnePerLine.pl | java -jar /mnt/e/Bioinformatics/tools/snpeff/snpEff/SnpSift.jar extractFields - CHROM POS REF ALT QUAL DP ANN "ANN[].ALLELE" "ANN[].EFFECT" "ANN[].IMPACT:" "ANN[].GENE:" "ANN[].GENEID:" ANN[].FEATURE" "ANN[].FEATUREID" "ANN[].BIOTYPE:" "ANN[].RANK:" "ANN[].HGVS_C" "ANN[].HGVS_P" "ANN[].CDNA_POS" "ANN[].CDNA_LEN" "ANN[].CDS_POS" "ANN[].CDS_LEN" "ANN[].AA_POS" "ANN[].AA_LEN" "ANN[].DISTANCE" "ANN[*].ERRORS" > snpsiftout8.vcf \
I faced 2 issues:
1- the code hanged at > sign, for longer time than expected
2- I tried to shorten the parameters in order to see where is the hang, by using:
$ cat my.ann.vcf | /mnt/e/Bioinformatics/tools/snpeff/snpEff/scripts/vcfEffOnePerLine.pl |java -jar /mnt/e/Bioinformatics/tools/snpeff/snpEff/SnpSift.jar extractFields - CHROM POS REF ALT QUAL DP "ANN[].ALLELE" "ANN[].EFFECT" "ANN[].GENE:" "ANN[].GENEID:" > snpsiftout2.vcf
then I got:
line 1:11 no viable alternative at input '[].GENE:'
line 1:13 no viable alternative at input '[].GENEID:'
I appreciate any insights,
Thanks

Hi,
my vcf file has an info field named dbNSFP_pos(1-coor). When I try to extract it with extractFields it causes unknown function exception. How to escape parenthesis?
Thanks.
version:
SnpSift 4.3t (build 2017-11-24 10:18), by Pablo Cingolani
command:
SnpSift Annotate -info LEGACY_ID,CNT -name COSMIC_ /gene/reference/COSMIC/GRCh37/v90/CosmicCodingMuts.vcf.gz test.vcf
database lines:
11 1016694 COSV70141335 G A . . GENE=MUC6;STRAND=-;LEGACY_ID=COSM922168;CDS=c.6107C>T;AA=p.A2036V;CNT=1
11 1016704 COSV70132597 C A . . GENE=MUC6;STRAND=-;LEGACY_ID=COSM4145033;SNP;CDS=c.6097G>T;AA=p.A2033S;CNT=23
11 1016708 COSV70137254 C T . . GENE=MUC6;STRAND=-;LEGACY_ID=COSM5763387;CDS=c.6093G>A;AA=p.Q2031=;CNT=2
11 1016713 COSV70132604 T C . . GENE=MUC6;STRAND=-;LEGACY_ID=COSM4590447;SNP;CDS=c.6088A>G;AA=p.T2030A;CNT=31
11 1016714 COSV70132213 G A . . GENE=MUC6;STRAND=-;LEGACY_ID=COSM2107691;CDS=c.6087C>T;AA=p.H2029=;CNT=5
11 1016715 COSV70132219 T C . . GENE=MUC6;STRAND=-;LEGACY_ID=COSM4183344;SNP;CDS=c.6086A>G;AA=p.H2029R;CNT=2
11 1016719 COSV70132229 T G . . GENE=MUC6;STRAND=-;LEGACY_ID=COSM3927641;SNP;CDS=c.6082A>C;AA=p.T2028P;CNT=3
vcf lines:
chr11 1016713 COSV70132604 TG CA,CG . clustered_events;map_qual;multiallelic CONTQ=66.0;DP=406;ECNT=4;GERMQ=93;MBQ=20,20,20;MFRL=0,0,0;MMQ=60,23,26;MPOS=27,28;NALOD=1.94,-55.0;NLOD=46.36,-29.23;POPAF=6.0,6.0;ROQ=93.0;SEQQ=88;STRANDQ=28;TLOD=13.6,13.6;COSMIC_CNT=31;COSMIC_LEGACY_ID=COSM4590447 GT:AD:AF:DP:F1R2:F2R1:SB 0/0:154,0,18:0.005714,0.109:172:73,0,9:73,0,9:80,74,14,4 0/1/2:222,6,6:0.03,0.03:234:103,2,3:103,2,3:94,128,8,4
it seems like, COSMIC only recorded the T -> C, but annotation is allele-free when there is a multi-allelic variant.
By the way, COSMIC may have multiple records at one chromosome coordinate, and SnpSift can annotate as a comma joint list correctly, but not the correct INFO with Number = 1 meta info;
SnpSift version 4.3t (build 2017-11-24 10:18)
When I tried to annotate my vcf using the gwas catalog, I found out that SnpSift gwasCat added the VCF header to the end of the my file.
Count homozygous ALT genotypes
SnpEffVersion="5.0e (build 2021-03-09 06:01)"
ANN[].Allele does not match with GEN[].GT of sample.
on the vcf file (attached as txt file), it shows:
#CHROM POS ID REF ALT FORMAT S1 S2 S3
Chr6 1789051 . CTTTTT CTTTT,C GT:AD:DP:GQ:PL 1/1:1,8,0:9:7:153,7,0,156,24,173 2/2:0,0,17:17:51:715,715,715,51,51,0 2/2:0,0,20:20:60:901,901,901,60,60,0
but on the file output of extractFields command, the output looks like this:
CHROM POS REF ALT GEN[S1].GT GEN[S2].GT GEN[S3].GT ANN[S1].ALLELE ANN[S2].ALLELE ANN[S3].ALLELE ANN[S1].IMPACT ANN[S2].IMPACT ANN[S3].IMPACT
Chr6 1789051 CTTTTT CTTTT,C 1/1 2/2 2/2 C C CTTTT MODIFIER MODIFIER MODIFIER
Hi
Would it be possible to add another parameter to SNPSIFT extractFields -ff FieldsFile to be able to pass fields as a text file (one field by line)?
Eventually, it will be great if two columns are provided in the file the second column will be the name of the column for that field.
Thanks
See email from Ömer Faruk Gerdan (March 19 2014).
java -jar SnpSift.jar annotate snpAnno.vcf sample.vcf > sample.anno.vcf
$ cat sample.vcf
1 884039 . C G 25.4 LowQual H=0.0311;A=0.0277 GT:AD:DP:GQ:PL ./.
1 884040 . A C 25.4 LowQual H=0.0311;A=0.0277 GT:AD:DP:GQ:PL ./.
1 884040 . AC A 334.27 PASS H=0.0323;A=0.0287 GT:AD:DP:GQ:PL ./.
$ cat snpAnno.vcf
1 884040 tr A C . . hetRatio=0.0323;homRatio=0.0287
1 884040 tr AC A . . hetRatio=0.1039;homRatio=0.0323
Compound Het filter
Hi @pcingola
When trying to annotate using SnpSift against local dbNSFP3.0.gz (dbNSFPv3.0b1a version)
Error: 'Uniprot_acc' field not found: I checked the data indeed not found ....how can SnpSift can ignore this?
Thanks for your support
00:00:00.000 SnpSift version 4.1h (build 2015-08-03), by Pablo Cingolani
00:00:00.006 Command: 'DBNSFP'
00:00:00.007 Reading configuration file 'snpEff.config'
00:00:00.315 done
00:00:00.316 Annotating
Input file : 'test.vcf'
Database file : '/data4/snpEff/data/dbNSFP3.0.gz'
00:00:00.442 Guessing data types
.................................00:00:11.737 Done
Error: Error: Field name 'Uniprot_acc' not found
Bug report:
....it would seem natural to replace the INFO lines for a tag that is being annotated with the one from the reference VCF used to annotate. Presently, it seems that these INFO lines are being left untouched and therefore you can end up potentially annotating a Number=A field into a file with a Number=1 header. Shouldn't snpEff always replace the header INFO lines?
Error: Unknown command 'annMem'
This is not working any more after commit 03c7cbb . Probably it should be just removed from the documentation?
The extract fields command does not recognize the ++ when I try to extract GERP++ scores from a VCF file.
Exception in thread "main" java.lang.RuntimeException: INFO field 'dbNSFP_GERP' not found in VCF header
at ca.mcgill.mcb.pcingola.snpSift.lang.expression.Field.getReturnType(Field.java:232)
at ca.mcgill.mcb.pcingola.snpSift.lang.expression.Field.eval(Field.java:56)
at ca.mcgill.mcb.pcingola.snpSift.SnpSiftCmdExtractFields.evaluate(SnpSiftCmdExtractFields.java:43)
at ca.mcgill.mcb.pcingola.snpSift.SnpSiftCmdExtractFields.run(SnpSiftCmdExtractFields.java:145)
at ca.mcgill.mcb.pcingola.snpSift.SnpSiftCmdExtractFields.run(SnpSiftCmdExtractFields.java:117)
at ca.mcgill.mcb.pcingola.snpSift.SnpSift.run(SnpSift.java:334)
at ca.mcgill.mcb.pcingola.snpSift.SnpSift.main(SnpSift.java:69)
From a user:
...sometimes one just wishes to know that a variant is present in a particular VCF
file (e.g. a blacklist of some sort) rather than pass on specific info about each variant..
Add a FLAG on whether a variant is present or not.
Apply VCF index methods to GFF, GTF, BED, etc.
Hi,
I have a vcf file annotated by snpeff and want to extract "ANN[*].RANK" from the ANN field like
ANN=G|missense_variant|MODERATE|TNN|ENSG00000120332|transcript|ENST00000239462|protein_coding|13/19|c.3038C>G|p.Thr1013Arg|3151/5008|3038/3900|1013/1299||
I used snpsift version 4.3t with the following command
java -jar /home/pmi/src/snpEff/SnpSift.jar extractFields -e "." my.vcf CHROM POS ID REF ALT "ANN[*].RANK"
but I got 13 not 13/19 for ANN[*].RANK.
thanks,
Hongen
Use "." on empty fields (or other character)
I am using SnpEFF 3.5 on human exomes with GRCh37.75 reference genome, and after I extract the fields I am interested in
java -jar /home/dcassate/snpEff/SnpSift.jar extractFields annotated_dbnsfp.vcf CHROM POS ID REF ALT EFF[].EFFECT EFF[].IMPACT EFF[].FUNCLASS EFF[].AA EFF[*].GENE dbNSFP_ESP6500_EA_AF dbNSFP_Interpro_domain dbNSFP_cds_strand > extracted.txt
I found that querying by, let's say, ESP6500 is not straightforward, as columns where data are missing are substituted by the next field.
Do you think there is a way to add a . or a - when the information for the specific field is absent?
Currently those fields are parsed as string.
Can anyone please send me the link to download dbSNP142.vcf file to be used to annotate my .vcf files.
I have a dbSNP142 file but its extension is .txt and not .vcf.
dbNSFP annotation seems to be inconsistently annotating the same line based on whether or not it is in the cache.
For testing purposes, created two files - a.vcf (two lines) and b.vcf (just the 2nd line). Annotated each file with dbNSFP, and the 2nd line gets different results - in the first file - no result, in the 2nd file - gets annotation.
a.vcf:
##fileformat=VCFv4.1 #CHROM POS ID REF ALT QUAL FILTER INFO chr2 102045328 . C T . PASS . chr2 102067083 . T C . PASS .
annotated with dbNSFP:
##fileformat=VCFv4.1 ##SnpSiftVersion="SnpSift 4.3t (build 2017-11-24 10:18), by Pablo Cingolani" ##SnpSiftCmd="SnpSift DbNsfp b.vcf" ##INFO=<ID=dbNSFP_CADD_phred,Number=A,Type=Float,Description="Field 'CADD_phred' from dbNSFP"> ##INFO=<ID=dbNSFP_Polyphen2_HDIV_pred,Number=A,Type=Character,Description="Field 'Polyphen2_HDIV_pred' from dbNSFP"> ##INFO=<ID=dbNSFP_GERP___RS,Number=A,Type=Float,Description="Field 'GERP++_RS' from dbNSFP"> ##INFO=<ID=dbNSFP_GERP___NR,Number=A,Type=Float,Description="Field 'GERP++_NR' from dbNSFP"> ##INFO=<ID=dbNSFP_MutationTaster_pred,Number=A,Type=Character,Description="Field 'MutationTaster_pred' from dbNSFP"> ##INFO=<ID=dbNSFP_Interpro_domain,Number=A,Type=String,Description="Field 'Interpro_domain' from dbNSFP"> ##INFO=<ID=dbNSFP_1000Gp1_AF,Number=A,Type=Float,Description="Field '1000Gp1_AF' from dbNSFP"> ##INFO=<ID=dbNSFP_ESP6500_AA_AF,Number=A,Type=Float,Description="Field 'ESP6500_AA_AF' from dbNSFP"> ##INFO=<ID=dbNSFP_Uniprot_acc,Number=A,Type=String,Description="Field 'Uniprot_acc' from dbNSFP"> ##INFO=<ID=dbNSFP_Polyphen2_HVAR_pred,Number=A,Type=Character,Description="Field 'Polyphen2_HVAR_pred' from dbNSFP"> ##INFO=<ID=dbNSFP_LRT_pred,Number=A,Type=Character,Description="Field 'LRT_pred' from dbNSFP"> ##INFO=<ID=dbNSFP_ESP6500_EA_AF,Number=A,Type=Float,Description="Field 'ESP6500_EA_AF' from dbNSFP"> #CHROM POS ID REF ALT QUAL FILTER INFO chr2 102045328 . C T . PASS . chr2 102067083 . T C . PASS .
b.vcf:
##fileformat=VCFv4.1 #CHROM POS ID REF ALT QUAL FILTER INFO chr2 102067083 . T C . PASS .
annotated with dbNSFP:
##fileformat=VCFv4.1 ##SnpSiftVersion="SnpSift 4.3t (build 2017-11-24 10:18), by Pablo Cingolani" ##SnpSiftCmd="SnpSift DbNsfp b.vcf" ##INFO=<ID=dbNSFP_CADD_phred,Number=A,Type=Float,Description="Field 'CADD_phred' from dbNSFP"> ##INFO=<ID=dbNSFP_Polyphen2_HDIV_pred,Number=A,Type=Character,Description="Field 'Polyphen2_HDIV_pred' from dbNSFP"> ##INFO=<ID=dbNSFP_GERP___RS,Number=A,Type=Float,Description="Field 'GERP++_RS' from dbNSFP"> ##INFO=<ID=dbNSFP_GERP___NR,Number=A,Type=Float,Description="Field 'GERP++_NR' from dbNSFP"> ##INFO=<ID=dbNSFP_MutationTaster_pred,Number=A,Type=Character,Description="Field 'MutationTaster_pred' from dbNSFP"> ##INFO=<ID=dbNSFP_Interpro_domain,Number=A,Type=String,Description="Field 'Interpro_domain' from dbNSFP"> ##INFO=<ID=dbNSFP_1000Gp1_AF,Number=A,Type=Float,Description="Field '1000Gp1_AF' from dbNSFP"> ##INFO=<ID=dbNSFP_ESP6500_AA_AF,Number=A,Type=Float,Description="Field 'ESP6500_AA_AF' from dbNSFP"> ##INFO=<ID=dbNSFP_Uniprot_acc,Number=A,Type=String,Description="Field 'Uniprot_acc' from dbNSFP"> ##INFO=<ID=dbNSFP_Polyphen2_HVAR_pred,Number=A,Type=Character,Description="Field 'Polyphen2_HVAR_pred' from dbNSFP"> ##INFO=<ID=dbNSFP_LRT_pred,Number=A,Type=Character,Description="Field 'LRT_pred' from dbNSFP"> ##INFO=<ID=dbNSFP_ESP6500_EA_AF,Number=A,Type=Float,Description="Field 'ESP6500_EA_AF' from dbNSFP"> #CHROM POS ID REF ALT QUAL FILTER INFO chr2 102067083 . T C . PASS dbNSFP_1000Gp1_AF=0.6927655677655677;dbNSFP_CADD_phred=15.50;dbNSFP_GERP___NR=4.54;dbNSFP_GERP___RS=0.712;dbNSFP_MutationTaster_pred=P,P
After removing caching from DbNsfp.java (block below), it becomes consistent again.
if (tiblocks.equals(latestTabixIteratorBlocks)) { // Used cached entries LinkedList<DbNsfpEntry> results = new LinkedList<>(); for (DbNsfpEntry de : latestResults) { if (match(variant, de)) results.add(de); else if (variant.getEnd() < de.getStart()) break; // Past query end? No need to continue. } return results; }
Expression based filtering (GTEX)
Hi Pablo,
I have a vcf file where some of the variants do not have for example the AD field. Then, trying to extract e.g. GEN[*].AD[0] leads to the following error:
Exception in thread "main" java.lang.NullPointerException
at ca.mcgill.mcb.pcingola.snpSift.lang.expression.FieldGenotypeSub.getFieldString(FieldGenotypeSub.java:29)
at ca.mcgill.mcb.pcingola.snpSift.SnpSiftCmdExtractFields.evaluate(SnpSiftCmdExtractFields.java:44)
at ca.mcgill.mcb.pcingola.snpSift.SnpSiftCmdExtractFields.run(SnpSiftCmdExtractFields.java:146)
at ca.mcgill.mcb.pcingola.snpSift.SnpSiftCmdExtractFields.run(SnpSiftCmdExtractFields.java:120)
at ca.mcgill.mcb.pcingola.snpSift.SnpSift.run(SnpSift.java:233)
at ca.mcgill.mcb.pcingola.snpSift.SnpSift.main(SnpSift.java:60)
I guess it would be nice if it was first checked whether the field actually exists for the variant to prevent this, what do you think?
Sometimes I will expect to conserve all genotypic calls with GQ>= 20, while changing the genotypes below the threshold to missings (./.).
Do you think snpSift already has such functionality?
A declarative, efficient, and flexible JavaScript library for building user interfaces.
🖖 Vue.js is a progressive, incrementally-adoptable JavaScript framework for building UI on the web.
TypeScript is a superset of JavaScript that compiles to clean JavaScript output.
An Open Source Machine Learning Framework for Everyone
The Web framework for perfectionists with deadlines.
A PHP framework for web artisans
Bring data to life with SVG, Canvas and HTML. 📊📈🎉
JavaScript (JS) is a lightweight interpreted programming language with first-class functions.
Some thing interesting about web. New door for the world.
A server is a program made to process requests and deliver data to clients.
Machine learning is a way of modeling and interpreting data that allows a piece of software to respond intelligently.
Some thing interesting about visualization, use data art
Some thing interesting about game, make everyone happy.
We are working to build community through open source technology. NB: members must have two-factor auth.
Open source projects and samples from Microsoft.
Google ❤️ Open Source for everyone.
Alibaba Open Source for everyone
Data-Driven Documents codes.
China tencent open source team.