Using scRNA-seq data, Asgard repurposes drugs and predicts personalized drug combinations to address the cellular heterogeneity of patients.
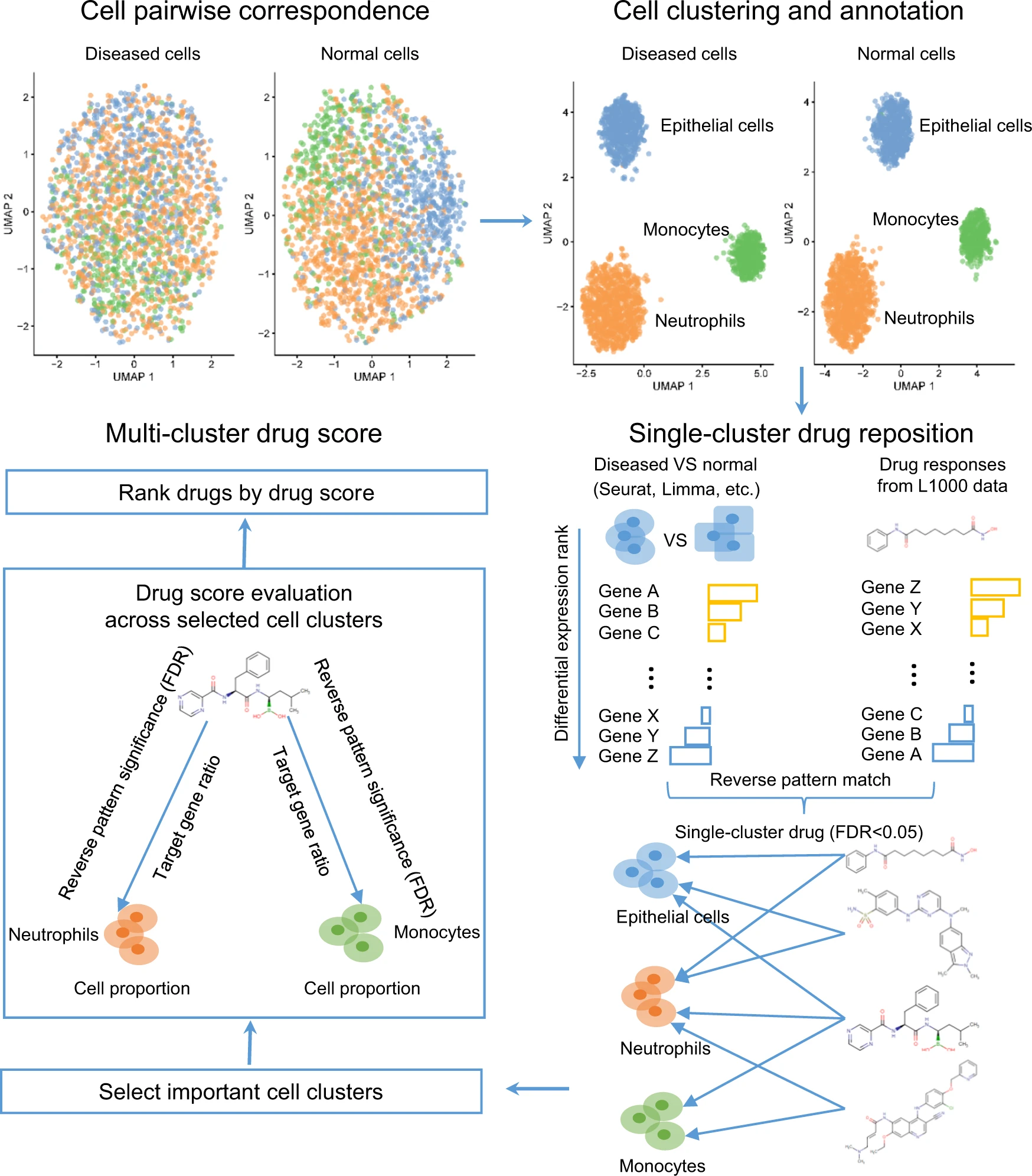
He, B., Xiao, Y., Liang, H. et al. ASGARD is A Single-cell Guided Pipeline to Aid Repurposing of Drugs. Nat Commun 14, 993 (2023). https://doi.org/10.1038/s41467-023-36637-3
Asgard package requires only a standard computer with enough RAM (>64GB) to support the in-memory operations.
The package has been tested on the following systems:
Windows 10
CentOS Linux 7
Seurat
limma
cmapR
SingleR
celldex
install.packages('devtools')
if (!requireNamespace("BiocManager", quietly = TRUE))
install.packages("BiocManager")
BiocManager::install(c("SingleR","limma","cmapR","celldex"))
install.packages('Seurat')
#If you can't install a package with above commands, try to download the gz file and install it locally.
#Take celldex package as an example:
#Downlaod the source package of celldex in linux
wget https://bioconductor.org/packages/release/data/experiment/src/contrib/celldex_1.0.0.tar.gz
#Start R
R
#Install celldex from the local source package
install.packages('celldex_1.0.0.tar.gz')
#Note: some dependency packages require R version newer than 4.0
devtools::install_github("lanagarmire/Asgard")
library('Asgard')
Method 1: click file names below
GSE70138_Broad_LINCS_cell_info_2017-04-28.txt
GSE70138_Broad_LINCS_Level5_COMPZ_n118050x12328_2017-03-06.gctx
GSE70138_Broad_LINCS_sig_info_2017-03-06.txt
GSE70138_Broad_LINCS_gene_info_2017-03-06.txt
GSE92742_Broad_LINCS_cell_info.txt
GSE92742_Broad_LINCS_Level5_COMPZ.MODZ_n473647x12328.gctx
GSE92742_Broad_LINCS_sig_info.txt
or Method 2: run following commands in linux
wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE70nnn/GSE70138/suppl/GSE70138_Broad_LINCS_cell_info_2017-04-28.txt.gz
wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE70nnn/GSE70138/suppl/GSE70138_Broad_LINCS_Level5_COMPZ_n118050x12328_2017-03-06.gctx.gz
wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE70nnn/GSE70138/suppl/GSE70138_Broad_LINCS_sig_info_2017-03-06.txt.gz
wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE70nnn/GSE70138/suppl/GSE70138_Broad_LINCS_gene_info_2017-03-06.txt.gz
wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE92nnn/GSE92742/suppl/GSE92742_Broad_LINCS_cell_info.txt.gz
wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE92nnn/GSE92742/suppl/GSE92742_Broad_LINCS_Level5_COMPZ.MODZ_n473647x12328.gctx.gz
wget https://ftp.ncbi.nlm.nih.gov/geo/series/GSE92nnn/GSE92742/suppl/GSE92742_Broad_LINCS_sig_info.txt.gz
Unzip downloaded files, revise the Your_local_path and run the following code:
library('Asgard')
#Please replace Your_local_path with your real local folder
PrepareReference(cell.info="GSE70138_Broad_LINCS_cell_info_2017-04-28.txt",
gene.info="GSE70138_Broad_LINCS_gene_info_2017-03-06.txt",
GSE70138.sig.info = "GSE70138_Broad_LINCS_sig_info_2017-03-06.txt",
GSE92742.sig.info = "GSE92742_Broad_LINCS_sig_info.txt",
GSE70138.gctx = "GSE70138_Broad_LINCS_Level5_COMPZ_n118050x12328_2017-03-06.gctx",
GSE92742.gctx = "GSE92742_Broad_LINCS_Level5_COMPZ.MODZ_n473647x12328.gctx",
Output.Dir = "DrugReference/"
)
#Note: the file names here maybe different after unzipping.
#Please note that it takes more than one hour to produce drug references in a standard computer with RAM>64GB.
Please use '?PrepareReference' for more help.
Download datasets GSE113197 and GSE123926 from GEO before running this script.
Human Breast Cancer Epithelial Cells (GSE123926): GSE123926_RAW.tar
Normal Human Breast Epithelial Cells (GSE113197): GSE113197_RAW.tar
library('Seurat')
#Load normal sample Ind5 from GSE113197 dataset
celltype<-read.table(file="https://raw.githubusercontent.com/lanagarmire/Single-cell-drug-repositioning/master/Normal_celltype.txt",header = T,check.names=FALSE)
data<-read.table(file="GSM3099847_Ind5_Expression_Matrix.txt",header = T,check.names=FALSE)
row.names(data)<-data[,1]
data<-data[,-1]
celltype2<-subset(celltype,sample=="Ind5" & celltype %in% c("Luminal_L2_epithelial_cells","Luminal_L1.1_epithelial_cells", "Luminal_L1.2_epithelial_cells", "Basal_epithelial_cells"))
common <- intersect(colnames(data), rownames(celltype2))
data<-data[,common]
Epithelial2 <- CreateSeuratObject(counts = data, project = "Epithelial", min.cells = 3, min.features = 200,meta.data=data.frame(celltype2,cell=colnames(data),type="Normal"))
#Load normal sample Ind6 from GSE113197 dataset
data<-read.table(file="GSM3099848_Ind6_Expression_Matrix.txt",header = T,check.names=FALSE)
row.names(data)<-data[,1]
data<-data[,-1]
celltype3<-subset(celltype,sample=="Ind6" & celltype %in% c("Luminal_L2_epithelial_cells","Luminal_L1.1_epithelial_cells", "Luminal_L1.2_epithelial_cells", "Basal_epithelial_cells"))
common <- intersect(colnames(data), rownames(celltype3))
data<-data[,common]
Epithelial3 <- CreateSeuratObject(counts = data, project = "Epithelial", min.cells = 3, min.features = 200,meta.data=data.frame(celltype3,cell=colnames(data),type="Normal"))
#Load normal sample Ind7 from GSE113197 dataset
data<-read.table(file="GSM3099849_Ind7_Expression_Matrix.txt",header = T,check.names=FALSE)
row.names(data)<-data[,1]
data<-data[,-1]
celltype4<-subset(celltype,sample=="Ind7" & celltype %in% c("Luminal_L2_epithelial_cells","Luminal_L1.1_epithelial_cells", "Luminal_L1.2_epithelial_cells", "Basal_epithelial_cells"))
common <- intersect(colnames(data), rownames(celltype4))
data<-data[,common]
Epithelial4 <- CreateSeuratObject(counts = data, project = "Epithelial", min.cells = 3, min.features = 200,meta.data=data.frame(celltype4,cell=colnames(data),type="Normal"))
#Load cancer sample PDX110 from GSE123926 dataset
TNBC_PDX.data<- Read10X(data.dir = "GSM3516947_PDX110")
TNBC.PDX2 <- CreateSeuratObject(counts = TNBC_PDX.data, project = "TNBC", min.cells = 3, min.features = 200, meta.data=data.frame(row.names=colnames(TNBC_PDX.data), cell=colnames(TNBC_PDX.data), sample="PDX-110",type="TNBC.PDX"))
#Load cancer sample PDX322 from GSE123926 dataset
TNBC_PDX.data<- Read10X(data.dir = "GSM3516948_PDX322")
TNBC.PDX3 <- CreateSeuratObject(counts = TNBC_PDX.data, project = "TNBC", min.cells = 3, min.features = 200, meta.data=data.frame(row.names=colnames(TNBC_PDX.data), cell=colnames(TNBC_PDX.data), sample="PDX-332",type="TNBC.PDX"))
SC.list<-list(TNBC.PDX2=TNBC.PDX2,TNBC.PDX3=TNBC.PDX3,Epithelial2=Epithelial2,Epithelial3=Epithelial3,Epithelial4=Epithelial4)
CellCycle=TRUE #Set it TRUE if you want to do Cell Cycle Regression
anchor.features=2000
for (i in 1:length(SC.list)) {
SC.list[[i]] <- NormalizeData(SC.list[[i]], verbose = FALSE)
SC.list[[i]] <- FindVariableFeatures(SC.list[[i]], selection.method = "vst",
nfeatures = anchor.features, verbose = FALSE)
}
SC.anchors <- FindIntegrationAnchors(object.list = SC.list,anchor.features = anchor.features, dims = 1:15)
SC.integrated <- IntegrateData(anchorset = SC.anchors, dims = 1:15)
DefaultAssay(SC.integrated) <- "integrated"
if(CellCycle){
##Cell Cycle Regression
s.genes <- cc.genes$s.genes
g2m.genes <- cc.genes$g2m.genes
SC.integrated <- CellCycleScoring(SC.integrated, s.features = s.genes, g2m.features = g2m.genes, set.ident = TRUE)
SC.integrated <- ScaleData(SC.integrated, vars.to.regress = c("S.Score", "G2M.Score"), features = rownames(SC.integrated))
SC.integrated <- RunPCA(SC.integrated, npcs = 15, verbose = FALSE)
}else{
##Run the standard workflow for visualization and clustering
SC.integrated <- ScaleData(SC.integrated, verbose = FALSE)
SC.integrated <- RunPCA(SC.integrated, npcs = 15, verbose = FALSE)
}
##t-SNE and Clustering
SC.integrated <- RunUMAP(SC.integrated, reduction = "pca", dims = 1:15)
SC.integrated <- FindNeighbors(SC.integrated, reduction = "pca", dims = 1:15)
SC.integrated <- FindClusters(SC.integrated, algorithm = 1, resolution = 0.4)
##Cell Type Annotation, set by.CellType=TRUE if you want to annotate cell type.
by.CellType=FALSE
if(by.CellType == TRUE){
data <- as.matrix(SC.integrated@assays$RNA@data)
hpca.se <- HumanPrimaryCellAtlasData()
pred.hpca <- SingleR(test = data, ref = hpca.se, assay.type.test=1, labels = hpca.se$label.main)
cell.label <- data.frame(row.names = row.names(pred.hpca),celltype=pred.hpca$labels)
if(length([email protected]$celltype)>0){
[email protected]$celltype <- cell.label$celltype
}else{
[email protected] <- cbind([email protected],cell.label)
}
new.cells <- data.frame()
for(i in unique(SC.integrated$seurat_clusters)){
sub.data <- subset(SC.integrated,seurat_clusters==i)
temp <- table([email protected]$celltype)
best.cell <- names(which(temp==temp[which.max(temp)]))
cells.temp <- data.frame(cell.id=row.names([email protected]),celltype=best.cell)
new.cells <- rbind(new.cells,cells.temp)
}
cell.meta <- [email protected]
cell.id <- rownames(cell.meta)
row.names(new.cells) <- new.cells[,1]
new.cells <- new.cells[cell.id,]
[email protected]$celltype <- new.cells$celltype
}else{
[email protected]$celltype <- paste0("C",as.numeric([email protected]$seurat_clusters))
}
#Change sample names
sample<[email protected]$sample
sample[which(sample=="Ind5")]<-"Normal1"
sample[which(sample=="Ind6")]<-"Normal2"
sample[which(sample=="Ind7")]<-"Normal3"
[email protected]$sample<-sample
#Visualize alignment result
DimPlot(SC.integrated, reduction = "umap", split.by = "sample",group.by = "celltype")
#Case sample names
Case=c("PDX-110","PDX-332")
#Control sample names
Control=c("Normal1","Normal2","Normal3")
#Get differential gene expression profiles for every cell type (or cluster if without annotation) from Limma
library('limma')
DefaultAssay(SC.integrated) <- "RNA"
set.seed(123456)
Gene.list <- list()
C_names <- NULL
for(i in unique([email protected]$celltype)){
Idents(SC.integrated) <- "celltype"
c_cells <- subset(SC.integrated, celltype == i)
Idents(c_cells) <- "type"
[email protected]
Controlsample <- row.names(subset(Samples,sample %in% Control))
Casesample <- row.names(subset(Samples,sample %in% Case))
if(length(Controlsample)>min.cells & length(Casesample)>min.cells){
expr <- as.matrix(c_cells@assays$RNA@data)
new_expr <- as.matrix(expr[,c(Casesample,Controlsample)])
new_sample <- data.frame(Samples=c(Casesample,Controlsample),type=c(rep("Case",length(Casesample)),rep("Control",length(Controlsample))))
row.names(new_sample) <- paste(new_sample$Samples,row.names(new_sample),sep="_")
expr <- new_expr
bad <- which(rowSums(expr>0)<3)
expr <- expr[-bad,]
mm <- model.matrix(~0 + type, data = new_sample)
fit <- lmFit(expr, mm)
contr <- makeContrasts(typeCase - typeControl, levels = colnames(coef(fit)))
tmp <- contrasts.fit(fit, contrasts = contr)
tmp <- eBayes(tmp)
C_data <- topTable(tmp, sort.by = "P",n = nrow(tmp))
C_data_for_drug <- data.frame(row.names=row.names(C_data),score=C_data$t,adj.P.Val=C_data$adj.P.Val,P.Value=C_data$P.Value)
Gene.list[[i]] <- C_data_for_drug
C_names <- c(C_names,i)
}
}
names(Gene.list) <- C_names
#Get differential genes from Seurat (Wilcoxon Rank Sum test)
library('Seurat')
DefaultAssay(SC.integrated) <- "RNA"
set.seed(123456)
Gene.list <- list()
C_names <- NULL
for(i in unique([email protected]$celltype)){
Idents(SC.integrated) <- "celltype"
c_cells <- subset(SC.integrated, celltype == i)
Idents(c_cells) <- "type"
C_data <- FindMarkers(c_cells, ident.1 = "TNBC.PDX", ident.2 = "Normal")
C_data_for_drug <- data.frame(row.names=row.names(C_data),score=C_data$avg_logFC,adj.P.Val=C_data$p_val_adj,P.Value=C_data$p_val) ##for Seurat version > 4.0, please use avg_log2FC instead of avg_logFC
Gene.list[[i]] <- C_data_for_drug
C_names <- c(C_names,i)
}
names(Gene.list) <- C_names
#Get differential genes from DESeq2 method
library('Seurat')
DefaultAssay(SC.integrated) <- "RNA"
set.seed(123456)
Gene.list <- list()
C_names <- NULL
for(i in unique([email protected]$celltype)){
Idents(SC.integrated) <- "celltype"
c_cells <- subset(SC.integrated, celltype == i)
Idents(c_cells) <- "type"
C_data <- FindMarkers(c_cells, ident.1 = "TNBC.PDX", ident.2 = "Normal", test.use = "DESeq2")
C_data_for_drug <- data.frame(row.names=row.names(C_data),score=C_data$avg_logFC,adj.P.Val=C_data$p_val_adj,P.Value=C_data$p_val) ##for Seurat version > 4.0, please use avg_log2FC instead of avg_logFC
Gene.list[[i]] <- C_data_for_drug
C_names <- c(C_names,i)
}
names(Gene.list) <- C_names
#Get differential genes from EdgeR
library('edgeR')
Case=c("PDX-110","PDX-332")
Control=c("Normal1","Normal2","Normal3")
DefaultAssay(SC.integrated) <- "RNA"
set.seed(123456)
min.cells=3 # The minimum number of cells for a cell type. A cell type is omitted if it has less cells than the minimum number.
Gene.list <- list()
C_names <- NULL
for(i in unique([email protected]$celltype)){
Idents(SC.integrated) <- "celltype"
c_cells <- subset(SC.integrated, celltype == i)
Idents(c_cells) <- "type"
[email protected]
Controlsample <- row.names(subset(Samples,sample %in% Control))
Casesample <- row.names(subset(Samples,sample %in% Case))
if(length(Controlsample)>min.cells & length(Casesample)>min.cells){
expr <- as.matrix(c_cells@assays$RNA@data)
new_expr <- as.matrix(expr[,c(Casesample,Controlsample)])
new_sample <- data.frame(Samples=c(Casesample,Controlsample),type=c(rep("Case",length(Casesample)),rep("Control",length(Controlsample))))
row.names(new_sample) <- paste(new_sample$Samples,row.names(new_sample),sep="_")
expr <- new_expr
bad <- which(rowSums(expr>0)<3)
expr <- expr[-bad,]
group <- new_sample$type
dge <- DGEList(counts=expr, group=group)
group_edgeR <- factor(group,levels = c("Control","Case"))
design <- model.matrix(~ group_edgeR)
dge <- estimateDisp(dge, design = design)
fit <- glmFit(dge, design)
res <- glmLRT(fit)
C_data <- res$table
C_data_for_drug <- data.frame(row.names=row.names(C_data),score=C_data$logFC,adj.P.Val=p.adjust(C_data$PValue,method = "BH"),P.Value=C_data$PValue)
Gene.list[[i]] <- C_data_for_drug
C_names <- c(C_names,i)
}
}
names(Gene.list) <- C_names
library('Asgard')
#Load tissue specific drug reference produced by PrepareReference function as mentioned above. Please select proper tissue accroding to the disease.
my_gene_info<-read.table(file="DrugReference/breast_gene_info.txt",sep="\t",header = T,quote = "")
my_drug_info<-read.table(file="DrugReference/breast_drug_info.txt",sep="\t",header = T,quote = "")
drug.ref.profiles = GetDrugRef(drug.response.path = 'DrugReference/breast_rankMatrix.txt',
probe.to.genes = my_gene_info,
drug.info = my_drug_info)
#Repurpose mono-drugs for every cell type
Drug.ident.res = GetDrug(gene.data = Gene.list,
drug.ref.profiles = drug.ref.profiles,
repurposing.unit = "drug",
connectivity = "negative",
drug.type = "FDA")
Use '?GetDrug' for more help
library('Asgard')
library('Seurat')
GSE92742.gctx.path="GSE92742_Broad_LINCS_Level5_COMPZ.MODZ_n473647x12328.gctx"
GSE70138.gctx.path="GSE70138_Broad_LINCS_Level5_COMPZ_n118050x12328_2017-03-06.gctx
Tissue="breast"
Drug.score<-DrugScore(SC.integrated=SC.integrated,
Gene.data=Gene.list,
Cell.type=NULL,
Drug.data=Drug.ident.res,
FDA.drug.only=TRUE,
Case=Case,
Tissue="breast",
GSE92742.gctx=GSE92742.gctx.path,
GSE70138.gctx=GSE70138.gctx.path)
#Cell.type: select cell types/clusters to be used for drug score estimation
#Case: select samples to be used for drug score estimation
#Please use " " instead of "-" in tissue name, for example, while haematopoietic-and-lymphoid-tissue is the prefix of the drug reference files, the corresponding tissue name is "haematopoietic and lymphoid tissue".
Use '?DrugScore' for more help
library('Asgard')
library('Seurat')
#Select drug using drug socre
library(Hmisc)
Final.drugs<-subset(Drug.score,Drug.therapeutic.score>quantile(Drug.score$Drug.therapeutic.score, 0.99,na.rm=T) & FDR <0.05)
#Select drug for individual clusters
Final.drugs<-TopDrug(SC.integrated=SC.integrated,
Drug.data=Drug.ident.res,
Drug.FDR=0.1,
FDA.drug.only=TRUE,
Case=Case.samples,
DrugScore=FALSE
)
library('Asgard')
library('Seurat')
GSE92742.gctx.path="GSE92742_Broad_LINCS_Level5_COMPZ.MODZ_n473647x12328.gctx"
GSE70138.gctx.path="GSE70138_Broad_LINCS_Level5_COMPZ_n118050x12328_2017-03-06.gctx"
Drug.combinations<-DrugCombination(SC.integrated=SC.integrated,
Gene.data=Gene.list,
Drug.data=Drug.ident.res,
Drug.FDR=0.1,
FDA.drug.only=TRUE,
Combined.drugs=2,
Case=Case,
Tissue="breast",
GSE92742.gctx=GSE92742.gctx.path,
GSE70138.gctx=GSE70138.gctx.path)
Please use '?DrugCombination' for more help.
library('Asgard')
Final.combinations<-TopCombination(Drug.combination=Drug.combinations,
Combination.FDR=0.1,
Min.combination.score=1
)
Demo codes using real datasets are available at: https://github.com/lanagarmire/Single-cell-drug-repositioning
If you have further questions or comments, please contact Dr.Bing He: [email protected] or [email protected]

