
Drug Discovery has become more and more expensive over the years. One of the most challenging parts is finding candidate drug molecules that will be optimized in later stages. While drug discovery is becoming more challenging, at the same time, datasets related to drug discovery are continually increasing in size and are now more accesible to the public. Examples include;
To reduce the cost of drug discovery, we can apply big data techniques to mine these chemical databases and find new uses for old drug molecules, underutilized protein targets, or novel pathways for diseases.
To help in mining these unstructured and messy datasets, I have created a python package to translate these datasets into drug interaction networks which act as graph databases. Using these graph databases, medicinal chemists can quickly search, filter, and prepare subsets of the drug interaction network related to their study. These smaller datasets can be used for machine learning models, visualizations, and property anlaysis. Drug interaction networks stored in graph databases will help streamline the drug discovery process by making the tedious task of dataset preparation much more simpler and user friendly.
Graph data structures provide for a way to store and represent complex, heterogenous data sources. Unlike other data structures, graphs focuses on relationships between objects. This is different from relational data models (SQL) where relationships between two different objects have to be inferred by the use of foreign keys to perform complex joins. For applications that require quick lookup of long-distance interactions, graph data structures provide an improvement over relational data models.
Graph data structures are naturally well-suited to represent and store the complex relationships between drugs/compounds their protein targets and their associated diseases.
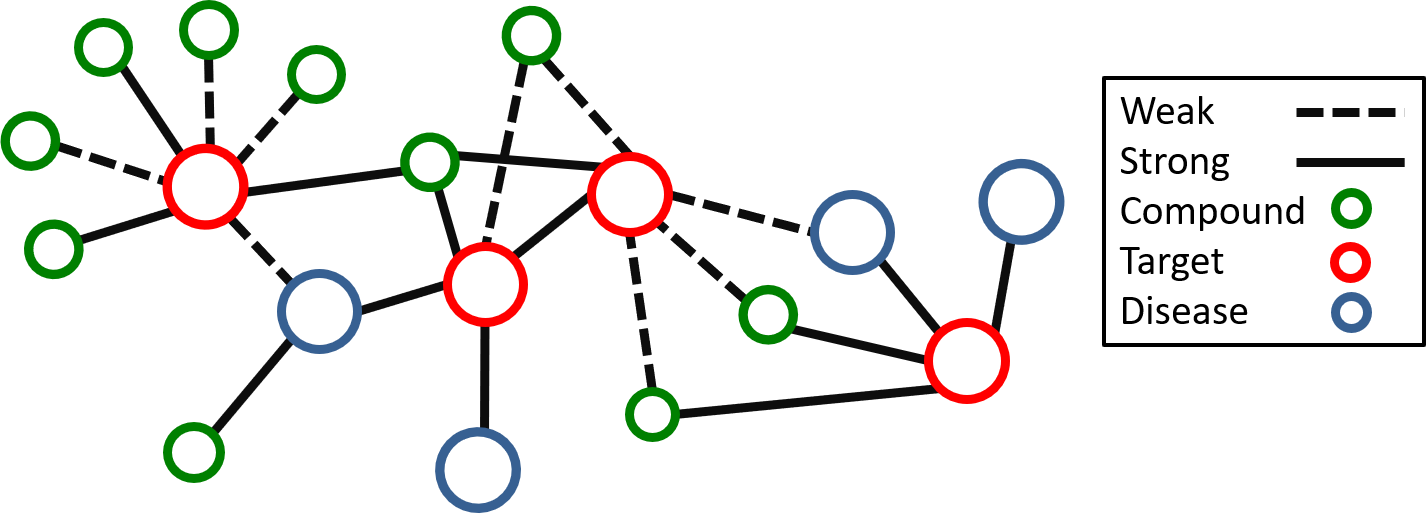
Aside from a convenient way to represent and store complex data, multiple algorithms have been developed to study graphs. These methods include graph traversal methods (BFS, DFS, Dijkstra's algorithm etc...) for searching specific relationships between objects in the graphs, community detection (Girvan-Newman Algorithm), clustering, label-propagation, and anomaly detection. These methods are already being used in drug discovery. For example anomaly detection of drug-target networks can be used to identify drug molecules that may display polypharmacological activity. Community detection and clustering can be used to identify groups of protein targets that are strongly associated with a disease. Label propagation can be used as a semi-supervised learning approach to predict drug and side-effect interactions. Since graph data structures already store relationships between drugs/compounds, protein targets, and diseases, graphs can be used as a graph database that can be used to quickly retrieve select protein targets and the compounds that bind to them. Graph-databases like Neo4j have already been applied to store data related to drug discovery. Finally graphs can be used to easily and clerly visualize the complex relationships of the drug/target network.
This repo describes my work in applying graph and network based methods to drug discovery. Using data from public sources like the ChemBL, MedDRA, UniProt, and OMIM, we can store the complex relationships between these different data sources as a graph database and apply the methods mentioned above to help advance drug discovery.
I have prepared tutorials in jupyter notebooks that will describe some techniques and applications of graph network.
-
How to extract data from the ChemBL 24 Postgresql database using psycopg2 and save the protein-ligand interactions in a networkx multigraph. The ChemBL 24 database has a lot of complex information related to protein-ligand assays as well as other information related to individual proteins. Dataset Creation
-
How to load the dataset and extract a smaller sub-set based on some selection criteria. Using this smaller sub-set we can generate a training/test set to train and validate a multi-label, random forest classification model that can predict which molecules will interact with which protein. Dataset Preperation
-
How to select and visualize the drugs that interact with a sub-set of protein targets. Visualization
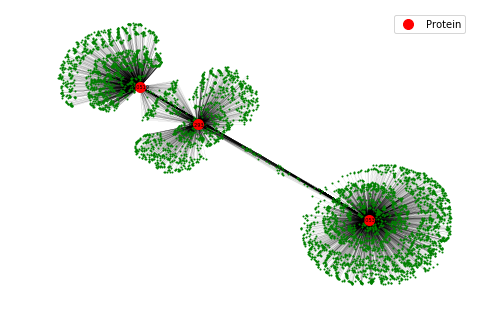
An example of the visualization is shown below. Here we see the drugs (green nodes) that interact with three target proteins (red nodes). The visualization can immediately reveal drug molecules that can simultaneously interact with more than one protein as shown by green nodes that have two edges connecting to two red nodes. These dual-targeting drugs are especially interesting and can be the starting point of a drug discovery campaign.
python 2.7networkxnumpyrdkitscipysklearn

