
An R package for analysis of data produced by transcript 5' profiling methods like RAMPAGE and MAPCap.
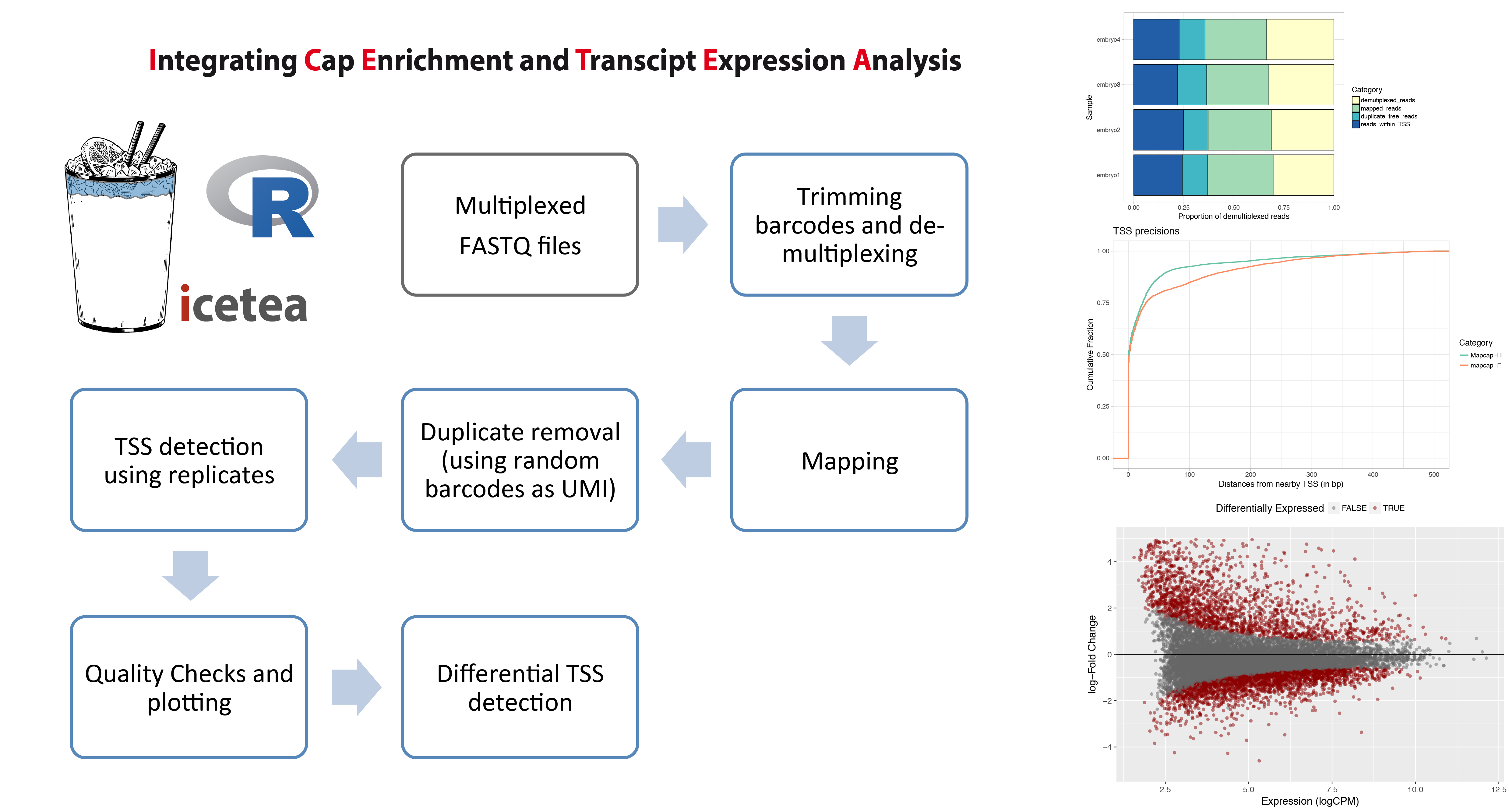
The icetea R package for analysis of TSS profiling data allows users to processes data from multiplexed, 5’-profiling techniques such as RAMPAGE and the recently developed MAPCap protocol. TSS detection and differential TSS analysis can be performed using replicates on any 5’-profiling dataset. Left panel : Typical analysis steps for MAPCap data that can be performed using icetea. Right panel : Showing some of the quality control and visualization outputs from the package. Proportion of sequencing reads used for each step (Top), comparison of TSS accuracy (w.r.t. annotated TSS) between samples (middle), and MA-plots from differential TSS analysis (Bottom).
Additionally, analysis of RNA-binding protein locations via the FLASH protocol can also be preformed via icetea.
Stable release of icetea is available via bioconductor.
## first install BiocManager
install.packages("BiocManager")
## then install icetea
BiocManager::install("icetea")
The latest (development) version of icetea can be installed using Devtools. Devtools can be installed from CRAN.
## first install devtools
install.packages("devtools")
## then install icetea
devtools::install_github("vivekbhr/icetea")
Please visit the icetea website for the package documentation and vignette.
If you use icetea in your analysis, please cite:
Bhardwaj, V., Semplicio, G., Erdogdu, N. U., Manke, T. & Akhtar, A. MAPCap allows high-resolution detection and differential expression analysis of transcription start sites. Nat. Commun. 10, 3219 (2019)






