My general text mining and text manipulation Python scripts.
Description of each script
find_overlap_in_lists_with_Venn.py
lists --> list of shared items and a diagram showing relationships between lists
Like find_overlap_in_lists.py (see below), except for in the cases of comparisons involving 2 or 3 list documents, it produces an area-weighted Venn diagram (image file) depicting the relationships of the items in the lists (or is it a Euler diagram?).
For comparing 4 lists, a Venn diagram will also be generated but it will not be area-weighted. To produce a Venn diagram for comparing lists found in four separate documents, it requires venn4_from_github.py (see below) in the same directory. This additional script is not needed in the cases comparing two or three lists.
The output files produced will include _shared_items and _overlap_ which can be used to easily track the newly created files, for example, after running the script you can run a move command to take advantage of this information to move the newly created files to a directory, such as mkdir new_data; mv *_shared_items* new_data/; mv *_overlap_* new_data/.
The script also sends to the command line some information about the percent matching the shared list. You can easily redirect that information to a file, like so python find_overlap_in_lists_with_Venn.py list1.txt list2.txt list3.txt 2> info.log. (The last part is saying to send the stderr, which is stream 2, to a file called info log.) Remember if you redirect the text, you won't see it in your teminal; check the new file.
See find_overlap_in_lists.py below for additional details.
Note: The core of this script and related code can produce a Venn diagram on mybinder.org-derived Binders if %matplotlib notebook or %matplotlib inline is invoked as the first line; you can find a demo here of that with the Venn diagram-producing portion.
Usage
usage: find_overlap_in_lists.py [-h] [-s] [-n] Files [Files ...]
find_overlap_in_lists.py takes any number of files of lists with items on
separate lines and reports those items shared by all. It was originally
intended to be used for lists of genes, but works on any lists. When comparing
2, 3, or four lists to each other, a Venn diagram depicting the relationship
of the lists will be made. Only the output for two or three lists will be an
area-weighted diagram.**** Script by Wayne Decatur (fomightez @ github) ***
positional arguments:
Files List the names of FILES containing the lists to compare. AT
LEAST ONE IS REQUIRED. Of course, it makes more sense with
two or more.
optional arguments:
-h, --help show this help message and exit
-s, --sensitive add this flag to force comparison of items to be case-
sensitive (NOT recommended). Default (recommended) is to
make the comparisons independent of character case in order
make matching more robust, and not miss matches when case
use is inconsistent among the lists.
-n, --nodiagram add this flag to force not making any Venn diagram. (Or you
could just use my script 'find_overlap_in_lists.py' that
does the same without producing a diagram.)
EXAMPLE OF INPUT AND OUTOUT for find_overlap_in_lists_with_Venn.py:
original input:
(text in three files with each column below representing contents of a file)
YDR190C YPR366C YDR112C
YPL235W YDr190C YdR190c
YLR366c YpL235W YPL235w
urgOu YLR466C YLR356C
SVLVASGYRHNITSVSQ urgou SVLVASGYRHNITSVSQ
YCL063W SVLVASGYRHNITSVSQ YFL037W
YML022W YBR845C YCL765C
YCR003W YDR772W YML022W
YIL007C YDL013W
YDL206W
YLR173W
YOR246C
YAL047C
YFL037W
YLR200W
YML124C
YER020C
YHL018W
YKL022C
YML100W
command:
python find_overlap_in_lists_with_Venn.py list1.txt list2.txt list3.txt
output after run:
(text in a file, called list1_and_2others_shared_items.txt , with the contents below)
YDR190C
YPL235W
SVLVASGYRHNITSVSQ
text shown on the command line (specifically, stderr):
Overlap identified! Shared items list saved as 'list1_and_2others_shared_items.txt'.
The overlap represents 30.00% of the list 'list1.txt'.
The overlap represents 15.00% of the list 'list2.txt'.
The overlap represents 37.50% of the list 'list3.txt'.
Image of overlap of the lists saved as 'list1_and_2others_overlap_representation.png'.
(image file called list1_and_2others_overlap_representation.png)

find_overlap_in_lists.py
lists --> list of shared items
Takes files with items listed one item to a line and determines items that occur in all the lists. Thus, it identifies only those items shared by all the lists of items. It was intended to be used for lists of genes, it but can work for any type of list.
A file listing the shared items (genes) will be produced.
The default settings cause the comparisons to be case-insensitive with the caveat that the case of the shared items listed in the output file will be determined by whatever list is provided in the first position of the file name list when calling the script.
Matching independent of case is done by default to make the comparisons more robust. The optional flag --sensitive can be used to override that behavior and make the comparisons case-sensitive. For case-sensitive matching, the order of the files when the script is called makes no difference as only perfect matches for all input lists will be in the output file.
Note, if using with yeast genes, probably best to use my script geneID_list_to_systematic_names.py, found in my yeastmine-related code repository to convert all to yeast systematic names so that the lists are standardized before running find_overlap_in_lists.py.
The output file(s) produced will include _shared_items which can be used to easily track the newly created files, for example, after running the script you can run a move command to take advantage of this information to move the newly created files to a directory, such as mkdir new_data; mv *_shared_items* new_data/.
The script also sends to the command line some information about the percent matching the shared list. You can easily redirect that information to a file, like so python find_overlap_in_lists.py list1.txt list2.txt list3.txt 2> info.log. The script also sends to the command line some information about the percent matching the shared list. You can easily redirect that information to a file, like so python find_overlap_in_lists_with_Venn.py list1.txt list2.txt list3.txt 2> info.log. (The last part is saying to send the stderr, which is stream 2, to a file called info log.) Remember if you redirect the text, you won't see it in your teminal; check the new file. Remember if you redirect the text, you won't see it in your teminal; check the new file.
A related script, find_overlap_in_lists_with_Venn.py, (see above) will do all this script will do plus when comparing 2, 3, or 4 lists it depicts the relationships among the lists in a Venn diagram (or is it a Euler diagram?).
Usage
usage: find_overlap_in_lists.py [-h] [-s] Files [Files ...]
find_overlap_in_lists.py takes any number of files of lists with items on
separate lines and reports those items shared by all. It was originally
intended to be used for lists of genes, but works on any lists. **** Script by
Wayne Decatur (fomightez @ github) ***
positional arguments:
Files Names of files containing lists to compare. AT LEAST ONE
REQUIRED.
optional arguments:
-h, --help show this help message and exit
-s, --sensitive add this flag to force comparison of items to be case-
sensitive (NOT recommended). Default (recommended) is to
make the comparisons independent of character case in order
make matching more robust, and not miss matches when case
use is inconsistent among the lists.
EXAMPLE OF INPUT AND OUTOUT for find_overlap_in_lists.py:
original input:
(text in three files with each column below representing contents of a file)
YDR190C YPR366C YDR112C
YPL235W YDr190C YdR190c
YLR366c YpL235W YPL235w
urgOu YLR466C YLR356C
SVLVASGYRHNITSVSQ urgou SVLVASGYRHNITSVSQ
SVLVASGYRHNITSVSQ
YBR845C
YDR772W
YDL013W
YDL206W
YLR173W
YOR246C
YAL047C
YFL037W
YLR200W
YML124C
command:
python find_overlap_in_lists.py list1.txt list2.txt list3.txt
output after run:
(text in a file, called list1_and_2others_shared_items.txt , with the contents below)
YDR190C
YPL235W
SVLVASGYRHNITSVSQ
text shown on the command line (specifically, stderr):
Overlap identified! Shared items list saved as 'list1_and_2others_shared_items.txt'.
The overlap represents 30.00% of the list 'list1.txt'.
The overlap represents 15.00% of the list 'list2.txt'.
The overlap represents 37.50% of the list 'list3.txt'.
extract_data_on_line_using_word_list.py
list of words and a data file --> lines of the data file that contain words in the provided list
Takes a file of words or names (could be gene identifiers, etc.) and then examines another file line by line and only keeps the lines in that file that contain the words or names the user provided.
The impetus for this to take a list of genes made using YeastMine and then extract lines from data on genes for just the provided list of genes. However, it is written more general than that to handle any sort of words and then to keep in a lines the "data file" that contain those words.
In the file that provides the word/name list, the list can be in almost any form, for example each word or name on a separate line or simply separated by a comma or orther punctuation or a mixture. By default spaces will be taken as the separation of words/names. If you'd like to specify that individual lines are the basic unit so that you can use more complex names or identifiers like "Mr. Smith", simply add the command line option --lines. Some attempt is made to even allow words like "don't" but it might not work for all cases such as the possesive forms of words ending in 's', like "Wiggins'". Punctuation here refers to any instances of these characters:
!"#$%&'()*+,-./:;<=>?@[]^_``{|}~
Matching is by default independent of case to make the comparisons more robust. The optional flag --sensitive can be used to override that behavior and make
the comparisons case-sensitive.
On the data side, you can specify that the words to match against only need to match a substring in the data lines in order to be kept by using the optional flag --data_substring_suffices. As an example, imagine the word list only contains the word "me". In the case of --data_substring_suffices a line with the word "some" on it will match and be kept. Whereas without the --data_substring_suffices flag, i.e., the default situation, only if the word "me" is on a line will the line be kept. This is useful for gene data because with it you can specify several genes. If you have "snR" as a word in your word list, you'd still get matches to lines containing "snR17" as well as lines containing "snR191".
The easiest way to run the script is to provide both the list of words or names file and the "data file" in the same directory with the script. However, if you are familiar with designating paths on the command line, thay can be used when invoking the script and pointing it at the files. The script will save the file in the same directory as the provided data file.
The easiest way to create a list_file using a YeastMine multi-column list is to paste it in a spreadsheet and extract the gene names column to a new file that you save as text. You'll want to use the --lines flag if working with tRNA genes like tP(UGG)A or any other identifier with punctuation.
Usage
usage: extract_data_on_line_using_word_list.py takes two files. One file will be used
as a list of words or names. That list will be used to examine line by line
the `data file` and lines containing words/names from the list will be kept
and placed in a resulting file. It was originally intended to be used for
lists of gene identifiers, but works with any words or names, etc. **** Script
by Wayne Decatur (fomightez @ github) ***
positional arguments:
ListFile Name of file containing list or words or names to look for
in lines of data file. REQUIRED.
DataFile Name of file containing lines to scan for the presence of
the provided list of words or names. Only those lines with
those words or names will be kept for the outout file.
REQUIRED.
optional arguments:
-h, --help show this help message and exit
-l, --lines add this flag to force individual lines to be used to
read the words_list and make the list to be compared
to lines in the data file. This enables the use of
two-word names with punctutation, like `Mr. Smith`, or
even phrases.
-d, --data_substring_suffices
add this flag to allow substrings from the data lines
to match contents of the words_list. For example, when
this flag is active `me` in the words_list would allow
for matches to lines containing the word `some`.
-s, --sensitive add this flag to force comparison of items to be case-
sensitive (NOT recommended). Default (recommended) is to
make the comparisons independent of character case in order
make matching more robust, and not miss matches when case
use is inconsistent among the sources.
EXAMPLE OF INPUT AND OUTOUT for extract_data_on_line_using_word_list.py:
original input for list_file:
(text in three files with each column below representing contents of a file)
YDR190C
YPL235W
YLR366c
urgOu
SVLVASGYRHNITSVSQCCTISSLRKVKVQLHCGGDRREELEIFTARACQCDMCRLSRY
original input for data_file:
(text in three files with each column below representing contents of a file)
YPR366C 87 7.91
YDr190C 99 6.54
YPL235W 78 32.21
YLR466C 17 7.91
urgou 54 9.87
SVLVASGYRHNITSVSQCCTISSLRKVKVQLHCGGDRREELEIFTARACQCDMCRLSRY 83 45.55
YBR845C 31 6.91
YDR772W 18 7.55
YDL013W 12 3.91
YDL206W 27 7.39
YLR173W 22 6.18
YOR246C 64 8.91
YAL047C 78 7.43
YFL037W 45 2.97
YLR200W 76 8.23
YML124C 88 3.51
YER020C 67 9.22
YHL018W 11 5.11
YKL022C 53 4.15
YML100W 91 8.62
command:
python extract_data_on_line_using_word_list.p list_file.txt data_file.txt
output after run:
(text in a file, called data_file_extracted.txt , with the contents below)
YPR366C 87 7.91
YDr190C 99 6.54
YPL235W 78 32.21
urgou 54 9.87
SVLVASGYRHNITSVSQCCTISSLRKVKVQLHCGGDRREELEIFTARACQCDMCRLSRY 83 45.55
text shown on the command line (specifically, stderr):
Extracted lines (5 total) saved as 'data_file_extracted.txt'.
subtract_data_on_line_using_word_list.py
list of words and a data file --> lines of the data file that contain words in the provided list
Takes a file of words or names (could be gene identifiers, etc.) and then examines another file line by line and only keeps the lines in that file that completely lack any of the words or names the user provided.
The impetus for this to take a list of genes made using YeastMine and then subtract lines from data on genes to remove the provided list of genes. However, it is written more general than that to handle any sort of words and then to remove lines from the "data file" that contain those words.
In the file that provides the word/name list, the list can be in almost any form, for example each word or name on a separate line or simply separated by a comma or orther punctuation or a mixture. By default spaces will be taken as the separation of words/names. If you'd like to specify that individual lines are the basic unit so that you can use more complex names or identifiers like "Mr. Smith", simply add the command line option --lines.
Some attempt is made to even allow words like "don't" but it might not work for all cases such as the possesive forms of words ending in 's', like "Wiggins'. Punctuation here refers to any instances of these characters:
!"#$%&'()*+,-./:;<=>?@[\]^_`{|}~
Matching is by default independent of case to make the comparisons more robust. The optional flag --sensitive can be used to override that behavior and make the comparisons case-sensitive.
On the data side, you can specify that the words to match against only need to match a substring in the data lines in order to be removed by using the optional flag --data_substring_suffices. As an example, imagine the word list only contains the word "me". In the case of --data_substring_suffices a line with the word "some" on it will match and be removed. Whereas without the --data_substring_suffices flag, i.e., the default situation, only if the word "me" is on a line will the line be removed This is useful for gene data because with it you can specify several genes. If you have "snR" as a word in your word list, you'd still get matches to lines containing "snR17" as well as lines containing "snR191".
The easiest way to run the script is to provide both the list of words or names file and the "data file" in the same directory with the script. However,if you are familiar with designating paths on the command line, thay can be used when invoking the script and pointing it at the files. The script will save the file in the same directory as the provided data file.
The easiest way to create a list_file using a YeastMine multi-column list is to paste it in a spreadsheet and extract the gene names column to a new file that you save as text. You'll want to use the --lines flag if working with tRNA genes like tP(UGG)A or any other identifier with punctuation.
Use this script if you are seeking the difference between two lists that are in file form, i.e., those line/items that occur in one file, but not the other without regard to specific line number; use as the 'data file' the file to remove lines from to produce the output and use as the 'word list' file the other file, which may have related line/items but that will simply be used for comparison to determine what to remove from the 'data file' and remain untouched. Another way to look at it, if you had list A and list B, using the script this way with list A as the data file will give you what is unique to list A relative list B, and is thus one part of the inverse operation of the script that identifies overlap; you could concatenate the results of doing that operation in both directions on two files to get the actual inverse of the overlap script, i.e., all items that don't overlap in both lists. As you are more likely to want to know what list specifically is missing something, I don't have a script that gives the exact inverse of the overlap of two lists.
Usage
usage: subtract_data_on_line_using_word_list.py [-h] [-l] [-s] ListFile DataFile
subtract_data_on_line_using_word_list.py takes two files. One file will be
used as a list of words or names. That list will be used to examine line by
line the `data file` and lines containing words/names from the list will be
removed. It was originally intended to be used for lists of gene identifiers,
but works with any words or names, etc. **** Script by Wayne Decatur
(fomightez @ github) ***
positional arguments:
ListFile Name of file containing list or words or names to look for
in lines of data file. REQUIRED.
DataFile Name of file containing lines to scan for the presence of
the provided list of words or names. Only those lines
withoyt any those words or names will be kept for the
outout file. REQUIRED.
optional arguments:
-h, --help show this help message and exit
-l, --lines add this flag to force individual lines to be used to read
the words_list and make the list to be compared to lines in
the data file. This enables the use of two-word names with
punctuation, like `Mr. Smith`, or even phrases.
-d, --data_substring_suffices
add this flag to allow substrings from the data lines
to match contents of the words_list. For example, when
this flag is active `me` in the words_list would allow
for matches to lines containing the word `some`.
-s, --sensitive add this flag to force comparison of items to be case-
sensitive (NOT recommended). Default (recommended) is to
make the comparisons independent of character case in order
make matching more robust, and not miss matches when case
use is inconsistent among the sources.
EXAMPLE OF INPUT AND OUTPUT for subtract_data_on_line_using_word_list.py with --data_substring_suffices flag used:
original input for list_file:
(text in three files with each column below representing contents of a file)
YDR190C
YPL235W
YPR366c
urgOu
SVLVASGYRHNITSVSQ
original input for data_file:
(text in three files with each column below representing contents of a file)
YPR366C 87 7.91
YDr190C 99 6.54
YPL235W 78 32.21
YLR466C 17 7.91
urgou 54 9.87
SVLVASGYRHNITSVSQCCTISSLRKVKVQLHCGGDRREELEIFTARACQCDMCRLSRY 83 45.55
YBR845C 31 6.91
YDR772W 18 7.55
YDL013W 12 3.91
YDL206W 27 7.39
command:
python subtract_data_on_line_using_word_list.p list_file.txt data_file.txt --data_substring_suffices
output after run:
(text in a file, called data_file_extracted.txt , with the contents below)
YLR466C 17 7.91
YBR845C 31 6.91
YDR772W 18 7.55
YDL013W 12 3.91
YDL206W 27 7.39
Note that SVLVASGYRHNITSVSQCCTISSLRKVKVQLHCGGDRREELEIFTARACQCDMCRLSRY 83 45.55 would have remained if --data_substring_suffices flag had not been included in the example command. The default is to need the word in word list to match entire word.
text shown on the command line (specifically, stderr):
Number of lines subtracted: 5.
Lines remaining saved as 'data_file_subtracted.txt'.
pull_MATCHING_data_from_files_in_directory.py
pertinent lines in several files --> all those lines in one file
pull_MATCHING_data_from_files_in_directory.py takes a directory as an argument and mines for lines in the file(s) matching the text of interest. It only mines the files that match constraints set for the file names. All the lines get saved to a file.
For the current version, the text of interest to match and the file name constraints are to be set as user-provided values in the code. Make sure to edit the code to match your needs.
Though it is currently configured to only focus on a directory of files, you can easily direct it at a single file by limiting the file name constraints to only match a single file and point the script at the directory that contains that file.
venn4_from_github.py
slightly modified from the original by Kristoffer Sahlin here to be used with
find_overlap_in_lists_with_Venn.py
df_subgroups_states2summary_df.py
dataframe / table of data --> summary data table with the ratio or percents for each subgrouping state per total and each group.
Typical input and result (the red annoation is just for illustration):
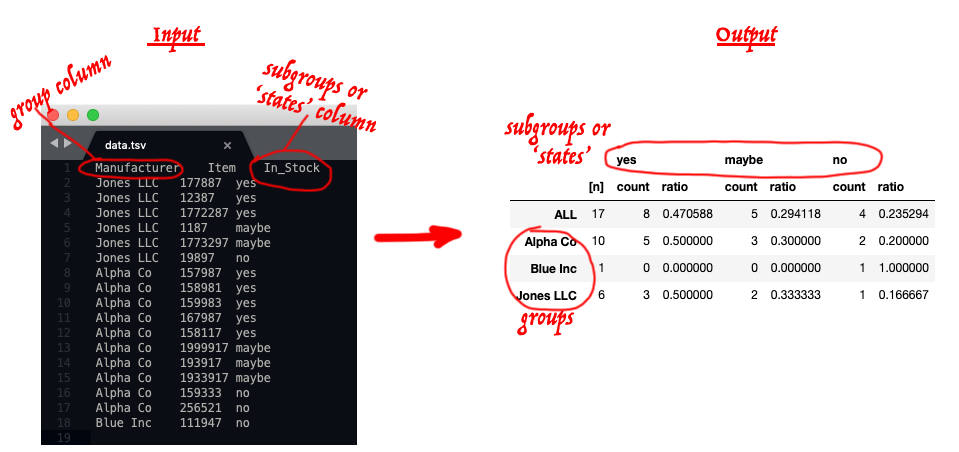
See here for more examples and to launch a demonstration session.
It's use is compared/contrasted with donut plots summarizing data here and here. Those notebooks can be launched in active form via the Binder service here. Depending on the formatting of the data table output the summary data table and the donut plot can both be complementary, too.
df_binary_states2summary_df.py
dataframe of data /table --> summary data table with the ratio or percents for the 'positive' state per total and each group.
Only works with binary states / subgroups because summarizes even further than df_subgroups_states2summary_df.py to just leave the 'positive' state and the other part is inferred by subtraction from 100%.
Typical input and result (the red annoation is just for illustration):
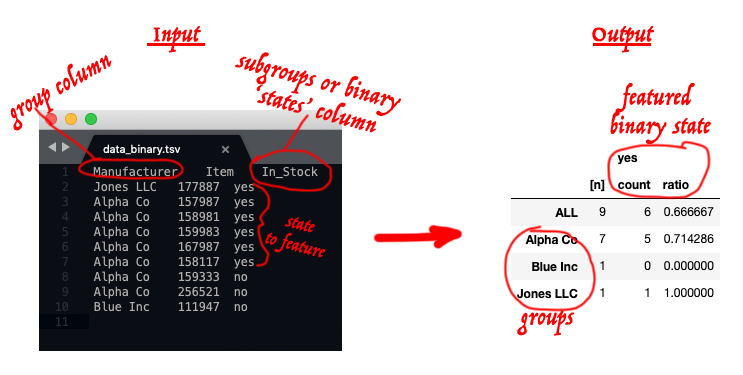
See here for more examples and to launch a demonstration session.
It's use is compared/contrasted with a donut plot summarizing data here. That notebook can be launched in active form via the Binder service here. Depending on the formatting of the data table output the summary data table and the donut plot can both be complementary, too.
csv_data_to_list.py
files with comma-separated values --> specified data from each file printed to stdout as a Python list
Presently a user-unfriendly utility script that needs the list of files hard-coded. Features a useful function csv_to_dict function takes a csv file and returns a dictionary where column headings (first line of th csv file) are mapped to a list of values for that column. The keys come from the first line of the csv file. Columns designated for the lists are alse set as hard-coded values in the script at this time.
function to generate accounting of positions in two ranked lists.py
two ranked lists --> information about how rows change between the two lists.
Presently a simple utility function to work in a Jupyter notebook.
Content & Arrangement of output inspired in a significant way by 'Rank list' accessible clicking Hide sample files and parameters [-] at Rank Rank Hypergeometric Overlap:Graeber Lab Homepage:Simple Version(RRHO).
See the documentation within the header of the script for more information. Alternatively, because a demonstration is often with more than a lot of words...
I made launchable notebook demonstrating its abilities with a list of yeast genes here. In developing it, I used my make_simulated_yeast_gene_set.py script to make a mock gene list.
collecting a column in tabbed input - specfically XXXXX.py(seems generalized ascolumn_tabbed_extraction.py) (Not here yet because needs further editing but drafted <-- seems old now and would need updating to my more recent script style)
TSV formatted table-like text (tab-delimited text) --> extracted column
info here
Several (most?) of my mini-pipeline scripts involve scripts that fall under this category.
Since data mining and text manipulations are at the heart of many of my computational endeavors, several other of my code repositories hold code related to text mining/text manipulation. Here are some:
-
- In particular,
compare_results_systematic_to_std.pyunder "Evaluation" sub-folder here is to highlight differences between a list with regard to whether each corresponding line is the same between both files. (Despite name, I think it is rather generally applicable. Or did I recognizediff, and/or for just a handful of total files, Text Wrangler, as good enough and didn't bother generalizing?) (If you are just looking to get difference between two lists that are in file form, i.e., those line/items that occur in one file, but not the other without regard to specific line number, you can use thesubtract_data_on_line_using_word_list.pyscript in this repo, and use as the 'data file' the file to remove lines from to produce the output and use as the 'word list' file the other file, which may have related line/items but that will simply be used for comparison to determine what to remove from the 'data file' and remain untouched. Another way to look at it, if you had list A and list B, using thesubtract_data_on_line_using_word_list.pyscript this way with list A as the data file will give you what is unique to list A relative list B, and is thus one part of the inverse operation of the script that identifies overlap; you could concatenate the results of doing that operation in both directions on two files to get the actual inverse of the overlap script, i.e., all items that don't overlap in both lists. As you are more likely to want to know specifically what list is missing something, I don't have a script that gives the exact inverse of the overlap of two lists.) - many others in that repo
- In particular,
-
- In particular,
tx2gene_generator.pyunderAdjust Listscollection, in general takes a list of items in first column of a text list in a designated file, skipping header line, and rerranges it to output. All in a concise and easily adapted form.
- In particular,
Handy, related utilities authored by others:
-
CSVtoTable - Simple, Python-based command-line utility to convert CSV files to searchable and sortable HTML table. (Note that it looks like editing the function
converthere it could be made to handle TSV files. -
For summarizing dataframes by 'exploration' approach, see pandas-profiling in action.
-
Use Vennerable in R, example with summarized data (like I have in my gist here) by Ming Tang at https://twitter.com/tangming2005/status/1141746845460258817
-
supervenn- "precise and easy-to-read multiple sets visualization in Python" -actually not Venn diagram producing but a visualization meant convey similar information data (has compariso to relate visualizations towards the bottom)
-
gget enrichr is a module that is part of gget. gget enrichr performs an enrichment analysis on a list of genes using Enrichr. (related code by me, see delineate_properties_of_yeast_snoRNAs_in_gene_list.py that checks for enrichment of various categories of yeast snoRNAs)
Venny - for quick-n-dirty because seems not area-weighted
See here for "a deep-learning based tool for information extraction in the biomedical domain".
** MEANT FOR MOVING TO ELSEWHERE **
I don't yet have a repository for this subject, and so putting this start of one here in the hopes I stumble upon this 'mining' info when needed:
Python Script to download hundreds of images from 'Google Images'. It is a ready-to-run code!
"if you are looking for a handy python script for 'searching' and 'downloading' hundreds of Google images to the local hard disk for you next DL project, that's the one:" - SOURCE:https://twitter.com/rasbt/status/982822969947848704 And one in reply uses "selenium to download images from google search".




