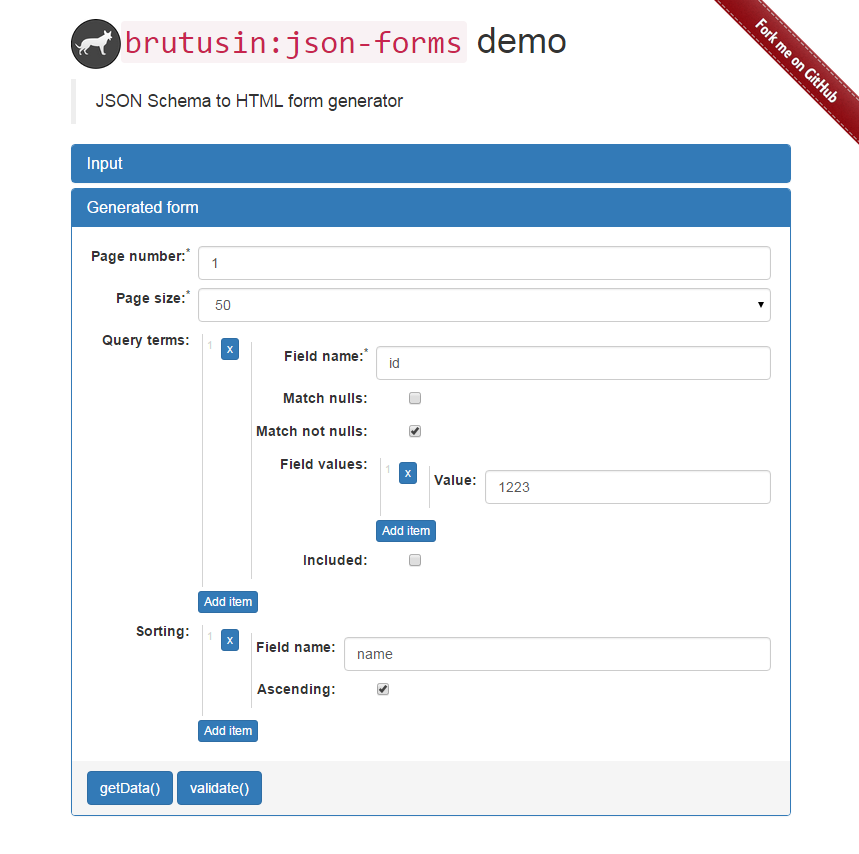
Current form's schema:
{"type": "object","name": "OPHTHALMOgenics Pipeline","description": "Pipeline aimed at helping the molecular diagnosis of cancer. It is aimed specifically at identifying all the genetic alterations that may influence susceptibility or resistance to approved drugs for clinical use . Pipeline analyzes DNA sequence of the genes and the presence/absence of clinically relevant rearrangements specified in selected probe.","properties": {"Parameters":{"description": "Pipeline execution global parameters","type": "object","properties": {"referenceGenomeVersion":{"description": ""Reference genome" (http://en.wikipedia.org/wiki/Reference_genome) version","required": true ,"title": "Reference Genome Version","default": "human/19/GRCh37","type":"string","enum":["human/19/GRCh37","human/19/LifeTechnologies","human/19/contigs","human/GRCh38/chr_contigs","human/GRCh38/chr_no_contigs","human/GRCh38/contigs","human/GRCh38/no_contigs","human/RNAseq/human/19/Ensembl/GRCh37/Annotation/NoPseudogenes","human/RNAseq/human/19/Ensembl/GRCh37/Annotation/SelectedBiotypes","human/RNAseq/human/19/Ensembl/GRCh37/Annotation/rRNA","human/RNAseq/human/19/Ensembl/GRCh37/Sequence/Bowtie2Index"]},"adapterSequences":{"description": "Adapter sequences used for DNA library generation in Trimmomatic (http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/TrimmomaticManual_V0.32.pdf) (fastaWithAdaptersEtc option)","required": true ,"title": "Adapter Sequences","default": "../param/adapters/Illumina/HiSeq-MiSeq/TruSeq3-PE.fa","type":"string","format":"dreampipe-file"},"reportProbeVersion":{"description": "Probe version (enrichment probes) used in bam statistics, callability and distance to probe annotation","required": false ,"title": "Report Probe Version","default": "OPHTHALMOgenics/V1-1","type":"string","enum":["OPHTHALMOgenics/V1","OPHTHALMOgenics/V1-1","OPHTHALMOgenics/V2_design","OPHTHALMOgenics/V2_report"]},"ensemblVersion":{"description": "Ensembl (http://www.ensembl.org/index.html) version","required": false ,"title": "Ensembl Version","default": "human/75-4","type":"string","enum":["ensembl/ensembl/human/75-2","ensembl/ensembl/human/79","ensembl/ensembl/human/BRCA1.BRCA2.TP53.v75","ensembl/human/75-2","ensembl/human/75-3","ensembl/human/75-4","ensembl/human/79","ensembl/human/BRCA1.BRCA2.TP53.v75","ensembl/human/BRCA1.BRCA2.TP53.v75-2","ensembl/mus-musculus/79-1","ensembl/saccharomyces-cerevisiae/75","human/72","human/75","human/75-1","human/75-2","human/75-3","human/75-4","human/79","human/BRCA1.BRCA2.TP53/V75_1","human/BRCA1.BRCA2.TP53/V75_2","mus-musculus/79","saccharomyces-cerevisiae/75"]},"distanceToProbeVersions":{"description": "Versions of the probes, used in the annotation step, for the calculation of the distance to probe","required": false ,"title": "Distance To Probe Versions","default": ["OPHTHALMOgenics/V1-1"],"type":"array","items":{"type":"string"},"enum":["Agilent/Agilent50MB","Agilent/Agilent50MB_extended200","Agilent/AgilentV4","Agilent/AgilentV4_UTR","Agilent/AgilentV5","Agilent/AgilentV5_UTR","Agilent/Agilent_SureSelect_ClinicalExome","Agilent/Agilent_humanallexon_V2_merged_extended200","Agilent/Agilent_humanallexon_V2_merged_nochr/data","Alejandro/data","AmpliSeqExome","HUCA/ITAV/V1","Haloplex_Clinic_v072013","ICGC_exome","ICGC_exome_extended200","IlluminaConfidentRegions","Illumina_ConfidentRegions_TruSight_One_V1_3","Illumina_TruSight_One/V1_1","Illumina_TruSight_One/V1_2","Illumina_TruSight_One/V1_3","Mouse_all_Exome/V1","NIST","Nimblegen/SeqCapExome/V3","OPHTHALMOgenics/V1","OPHTHALMOgenics/V1-1","OPHTHALMOgenics/V2_design","OPHTHALMOgenics/V2_report","OTOgenics/Homologs/V1","OTOgenics/OTOgenics/V1_design","OTOgenics/OTOgenics/V1_report","OTOgenics/OTOgenics/V2_design","OTOgenics/OTOgenics/V2_report","Oncogenics/Easy/V1","Oncogenics/Easy/V1_Homologs","Oncogenics/Easy/V1_covered","Oncogenics/Easy/V2_covered","Oncogenics/Easy/V5","Oncogenics/Germline/V4","Oncogenics/Germline/V5","Oncogenics/Germline/V5_report","Oncogenics/Homologous/Easy/V1","Oncogenics/Homologous/Germline/V1","Oncogenics/Oncogenics/V1_covered","Oncogenics/Oncogenics/V2_covered","Oncogenics/Oncogenics/V3_covered","Oncogenics/Oncogenics/V3_extended100","Oncogenics/Oncogenics/V3_extended50","Oncogenics/Oncogenics/V3_merged","Oncogenics/Oncogenics/V4_covered","Oncogenics/Oncogenics/V5","PMS2","Tusie_transcripts_BRCA1_BRCA2_TP53_EXTENDED/V1_0","colon_genes","dbSNP/144","nimblegen_SeqCap_EZ_Exome_v2_tiled","nimblegen_SeqCap_EZ_Exome_v2_tiled_extended200","uniovi/ONCOEPIC/V1"]},"positionGenomicDatabases":{"description": "Genomic databases used in annotation by position","required": false ,"title": "Position Genomic Databases","default": ["1000genomes/phase3-2","dbnsfp/3.0-1","cosmic/71-2","dbscSNV/20150107-1","dbsnp/all/142-2","dbsnp/clinvar/20150929-1","esp/6500-3","exac/0.3-2","hgmd/2015.1-3","icgc/19-1"],"type":"array","items":{"type":"string"},"enum":["1000genomes/phase3-1","1000genomes/phase3-2","bic/hg19","cll/20140217-3","cosmic/68-1","cosmic/71-1","cosmic/71-2","dbnsfp/2.8-2","dbnsfp/2.9-1","dbnsfp/3.0-1","dbscSNV/20150107-1","dbsnp/all/138-1","dbsnp/all/141-1","dbsnp/all/142-1","dbsnp/all/142-2","dbsnp/all/144-1","dbsnp/clinvar/20140303-1","dbsnp/clinvar/20150115-1","dbsnp/clinvar/20150305-1","dbsnp/clinvar/20150305-2","dbsnp/clinvar/20150629-1","dbsnp/clinvar/20150901-1","dbsnp/clinvar/20150929-1","dbsnp/common/137-1","dbsnp/oncogenicsV4/20150525-1","dgva/20140603-1","dlbcl-shm/20121012-3","esp/6500-1","esp/6500-2","esp/6500-3","exac/0.3-1","exac/0.3-2","hgmd/2014.1-2","hgmd/2014.1-3","hgmd/2015-2","hgmd/2015.1-3","icgc/17-1","icgc/18-1","icgc/19-1","repeat-masker/20140516-1","telcent/hg19-1","telcent/saccharomyces-cerevisiae/R64-1-1"]},"overlappingGenomicDatabases":{"description": "Genomic databases used in annotation by overlapping","required": false ,"title": "Overlapping Genomic Databases","default": ["pfam/20140516-1"],"type":"array","items":{"type":"string"},"enum":["1000genomes/phase3-1","bic/hg19","cll/20140217-3","cosmic/68-1","cosmic/71-1","dbnsfp/2.8-2","dbnsfp/2.9-1","dbsnp/all/138-1","dbsnp/all/141-1","dbsnp/all/142-1","dbsnp/all/144-1","dbsnp/clinvar/20140303-1","dbsnp/clinvar/20150115-1","dbsnp/clinvar/20150305-1","dbsnp/clinvar/20150305-2","dbsnp/clinvar/20150629-1","dbsnp/clinvar/20150901-1","dbsnp/common/137-1","dbsnp/oncogenicsV4/20150525-1","dgva/20140603-1","dlbcl-shm/20121012-3","esp/6500-1","esp/6500-2","exac/0.3-1","hgmd/2014.1-2","hgmd/2014.1-3","hgmd/2015-2","icgc/17-1","icgc/18-1","pfam/20140516-1","repeat-masker/20140516-1","telcent/hg19-1","telcent/saccharomyces-cerevisiae/R64-1-1"]},"minVarFreq":{"description": "Minimun mutation frequency (http://samtools.sourceforge.net/samtools.shtml)","required": true ,"title": "Min Var Freq","default": 0.15,"type":"number"},"md5ChecksumsFile":{"description": "One file containing all checksums for all input fastqs","required": false ,"title": "Md 5 Checksums File","type":"string","format":"dreampipe-file"},"threads":{"description": "Number of threads used in trimming and alignment","required": false ,"title": "Threads","default": 4,"type":"integer"},"mismatchesThreshold":{"description": "Maximum mismatches allowed in adapter in Trimmomatic (http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/TrimmomaticManual_V0.32.pdf) (seedMismatches option)","required": true ,"title": "Mismatches Threshold","default": 3,"type":"integer"},"palindromeThreshold":{"description": "Match accuracy between adapter-ligated reads in Trimmomatic (http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/TrimmomaticManual_V0.32.pdf) (palindromeClipThreshold option)","required": true ,"title": "Palindrome Threshold","default": 30,"type":"integer"},"simpleThreshold":{"description": "Match accuracy between adapter and read in Trimmomatic (http://www.usadellab.org/cms/uploads/supplementary/Trimmomatic/TrimmomaticManual_V0.32.pdf) (simpleClipThreshold option)","required": true ,"title": "Simple Threshold","default": 10,"type":"integer"},"headCrop":{"description": "commons.trimming.PairedEndScriptTask.headCrop","required": true ,"title": "Head Crop","default": 0,"type":"integer"},"aligner":{"description": "Aligner to be used. Choose between Bowtie2 (http://bowtie-bio.sourceforge.net/bowtie2/manual.shtml#what-is-bowtie-2), BWA (http://bio-bwa.sourceforge.net/bwa.shtml) or BWA_MEM (http://bio-bwa.sourceforge.net/bwa.shtml). Default is BWA_MEM","required": true ,"title": "Aligner","default": "bwa_mem","type":"string","enum":["bowtie2","bwa","bwa_mem"]},"gapOpen":{"description": "Maximum number of gap opens for aln command in BWA (http://bio-bwa.sourceforge.net/bwa.shtml) (-o option)","required": true ,"title": "Gap Open","default": 1,"type":"integer"},"mismatch":{"description": "Mismatch penalty for aln command in BWA (http://bio-bwa.sourceforge.net/bwa.shtml) (-M option)","required": true ,"title": "Mismatch","default": 3,"type":"integer"},"distance":{"description": "Fraction of allowed mismatches if the value is float, or maximum mismatches allowed if the value is an integer for aln command in BWA (http://bio-bwa.sourceforge.net/bwa.shtml) (-n option)","required": true ,"title": "Distance","default": 0.04,"type":"number"},"trim":{"description": "Minimum base quality for read trimming for aln command in BWA (http://bio-bwa.sourceforge.net/bwa.shtml) (-q option)","required": true ,"title": "Trim","default": 0,"type":"integer"},"clippingPenalty":{"description": "When performing SW extension, BWA-MEM keeps track of the best score reaching the end of query. If this score is larger than the best SW score minus the clipping penalty, clipping will not be applied.","required": false ,"title": "Clipping Penalty","default": 5,"type":"integer"},"homologousProbe":{"description": "Probe to use in homologous variant calling","required": false ,"title": "Homologous Probe","type":"string","enum":["Agilent/Agilent50MB","Agilent/Agilent50MB_extended200","Agilent/AgilentV4","Agilent/AgilentV4_UTR","Agilent/AgilentV5","Agilent/AgilentV5_UTR","Agilent/Agilent_SureSelect_ClinicalExome","Agilent/Agilent_humanallexon_V2_merged_extended200","Agilent/Agilent_humanallexon_V2_merged_nochr/data","Alejandro/data","AmpliSeqExome","HUCA/ITAV/V1","Haloplex_Clinic_v072013","ICGC_exome","ICGC_exome_extended200","IlluminaConfidentRegions","Illumina_ConfidentRegions_TruSight_One_V1_3","Illumina_TruSight_One/V1_1","Illumina_TruSight_One/V1_2","Illumina_TruSight_One/V1_3","Mouse_all_Exome/V1","NIST","Nimblegen/SeqCapExome/V3","OPHTHALMOgenics/V1","OPHTHALMOgenics/V1-1","OPHTHALMOgenics/V2_design","OPHTHALMOgenics/V2_report","OTOgenics/Homologs/V1","OTOgenics/OTOgenics/V1_design","OTOgenics/OTOgenics/V1_report","OTOgenics/OTOgenics/V2_design","OTOgenics/OTOgenics/V2_report","Oncogenics/Easy/V1","Oncogenics/Easy/V1_Homologs","Oncogenics/Easy/V1_covered","Oncogenics/Easy/V2_covered","Oncogenics/Easy/V5","Oncogenics/Germline/V4","Oncogenics/Germline/V5","Oncogenics/Germline/V5_report","Oncogenics/Homologous/Easy/V1","Oncogenics/Homologous/Germline/V1","Oncogenics/Oncogenics/V1_covered","Oncogenics/Oncogenics/V2_covered","Oncogenics/Oncogenics/V3_covered","Oncogenics/Oncogenics/V3_extended100","Oncogenics/Oncogenics/V3_extended50","Oncogenics/Oncogenics/V3_merged","Oncogenics/Oncogenics/V4_covered","Oncogenics/Oncogenics/V5","PMS2","Tusie_transcripts_BRCA1_BRCA2_TP53_EXTENDED/V1_0","colon_genes","dbSNP/144","nimblegen_SeqCap_EZ_Exome_v2_tiled","nimblegen_SeqCap_EZ_Exome_v2_tiled_extended200","uniovi/ONCOEPIC/V1"]},"rpkmFiles":{"description": "Set of rpkm files that will be used as background to do cnv calling ","required": false ,"title": "Rpkm Files","type":"array","items":{"type":"string"},"enum":["background","background_wey","tusie_rpkm_50_67","tusie_rpkm_samples","tusie_rpkm_samples_0","tusie_rpkm_samples_filtered","tusie_rpkm_samples_filtered_orginal","tussie_rpkm_23_49_filtered","ADN105_S1_ADN106_S2_rpkm.txt","ADN107_S1_rpkm.txt","HCE-10.P45_rpkm.txt","HCE-11.P46_rpkm.txt","HCE-12.P47_rpkm.txt","HCE-13.P48_rpkm.txt","HCE-14.P55_rpkm.txt","HCE-15.P58_rpkm.txt","HCE-16.P61_rpkm.txt","HCE-17.P3_rpkm.txt","HCE-18.P42_rpkm.txt","HCE-19.P44_rpkm.txt","HCE-20.P49E_rpkm.txt","HCE-21.P56_rpkm.txt","HCE-22.P60_rpkm.txt","HCE-23_filtered_rpkm.txt","HCE-24_filtered_rpkm.txt","HCE-25_filtered_rpkm.txt","HCE-26_filtered_rpkm.txt","HCE-27_filtered_rpkm.txt","HCE-28_filtered_rpkm.txt","HCE-29_filtered_rpkm.txt","HCE-30_filtered_rpkm.txt","HCE-31_filtered_rpkm.txt","HCE-32_filtered_rpkm.txt","HCE-33_filtered_rpkm.txt","HCE-34_filtered_rpkm.txt","HCE-35_filtered_rpkm.txt","HCE-36_filtered_rpkm.txt","HCE-37_filtered_rpkm.txt","HCE-38_filtered_rpkm.txt","HCE-39_filtered_rpkm.txt","HCE-40_filtered_rpkm.txt","HCE-41_filtered_rpkm.txt","HCE-42_filtered_rpkm.txt","HCE-43_filtered_rpkm.txt","HCE-44_filtered_rpkm.txt","HCE-45_filtered_rpkm.txt","HCE-46_filtered_rpkm.txt","HCE-47_filtered_rpkm.txt","HCE-48_filtered_rpkm.txt","HCE-49_filtered_rpkm.txt","HCE-50_rpkm.txt","HCE-51_rpkm.txt","HCE-52_rpkm.txt","HCE-53_rpkm.txt","HCE-54_rpkm.txt","HCE-55_rpkm.txt","HCE-56_rpkm.txt","HCE-57_rpkm.txt","HCE-58_rpkm.txt","HCE-59_rpkm.txt","HCE-6.P61_rpkm.txt","HCE-60_rpkm.txt","HCE-61_rpkm.txt","HCE-62_rpkm.txt","HCE-63_rpkm.txt","HCE-64_rpkm.txt","HCE-65_rpkm.txt","HCE-66_rpkm.txt","HCE-67_rpkm.txt","HCE-8.P4_rpkm.txt","HCE-9.P43_rpkm.txt","ONCOE.001_P01105_S1_rpkm.txt","ONCOE.001_P01105_filtered.txt","ONCOE.002_P01154_S1_rpkm.txt","ONCOE.002_P01154_filtered.txt","ONCOE.003_P01243_filtered.txt","ONCOE.004_P01273_filtered.txt","ONCOE.005_P01331_filtered.txt","ONCOE.006_P01351_filtered.txt","ONCOG.001_P01209_rpkm.txt","ONCOG.002_P01210_rpkm.txt","ONCOG.003_P01211_rpkm.txt","ONCOG.004_P01212_rpkm.txt","ONCOG.005_P01255_rpkm.txt","ONCOG.006_P00713_rpkm.txt","ONCOG.007_P00303_rpkm.txt","ONCOG.008_P00993_rpkm.txt","ONCOG.009_P01352.txt","ONCOG.009_P01352_rpkm.txt","ONCOG.010_P01256_filtered.txt","ONCOG.011_P01427_filtered.txt","ONCOG.012_P01440_filtered.txt","ONCOG.013_P01477_filtered.txt","P00303_rpkm.txt","P00492_S1_rpkm.txt","P00713_rpkm.txt","P00993_rpkm.txt","P01105_S1_rpkm.txt","P01154_S1_rpkm.txt","P01209_rpkm.txt","P01210_rpkm.txt","P01211_rpkm.txt","P01212_rpkm.txt","P01255_P01243_rpkm.txt","P01352.txt","pool_ADN105_S1_ADN106_S2_rpkm.txt","snp_array_ADN107_S1_rpkm.txt","snp_array_P00492_S1_rpkm.txt"]},"svd":{"description": "Number of SVD components to remove (zero). See below for more on this subject.","required": true ,"title": "Svd","default": 2,"type":"integer"},"threshold":{"description": "Threshold used in cnvs calling","required": true ,"title": "Threshold","default": 1,"type":"number"},"designProbeVersion":{"description": "Only if experiments design was ran with a different probe than the reports one, then this will be used in bams statistics, and annotation by distance to probe ","required": false ,"title": "Design Probe Version","default": "","type":"string","enum":["Agilent/Agilent50MB","Agilent/Agilent50MB_extended200","Agilent/AgilentV4","Agilent/AgilentV4_UTR","Agilent/AgilentV5","Agilent/AgilentV5_UTR","Agilent/Agilent_SureSelect_ClinicalExome","Agilent/Agilent_humanallexon_V2_merged_extended200","Agilent/Agilent_humanallexon_V2_merged_nochr/data","Alejandro/data","AmpliSeqExome","HUCA/ITAV/V1","Haloplex_Clinic_v072013","ICGC_exome","ICGC_exome_extended200","IlluminaConfidentRegions","Illumina_ConfidentRegions_TruSight_One_V1_3","Illumina_TruSight_One/V1_1","Illumina_TruSight_One/V1_2","Illumina_TruSight_One/V1_3","Mouse_all_Exome/V1","NIST","Nimblegen/SeqCapExome/V3","OPHTHALMOgenics/V1","OPHTHALMOgenics/V1-1","OPHTHALMOgenics/V2_design","OPHTHALMOgenics/V2_report","OTOgenics/Homologs/V1","OTOgenics/OTOgenics/V1_design","OTOgenics/OTOgenics/V1_report","OTOgenics/OTOgenics/V2_design","OTOgenics/OTOgenics/V2_report","Oncogenics/Easy/V1","Oncogenics/Easy/V1_Homologs","Oncogenics/Easy/V1_covered","Oncogenics/Easy/V2_covered","Oncogenics/Easy/V5","Oncogenics/Germline/V4","Oncogenics/Germline/V5","Oncogenics/Germline/V5_report","Oncogenics/Homologous/Easy/V1","Oncogenics/Homologous/Germline/V1","Oncogenics/Oncogenics/V1_covered","Oncogenics/Oncogenics/V2_covered","Oncogenics/Oncogenics/V3_covered","Oncogenics/Oncogenics/V3_extended100","Oncogenics/Oncogenics/V3_extended50","Oncogenics/Oncogenics/V3_merged","Oncogenics/Oncogenics/V4_covered","Oncogenics/Oncogenics/V5","PMS2","Tusie_transcripts_BRCA1_BRCA2_TP53_EXTENDED/V1_0","colon_genes","dbSNP/144","nimblegen_SeqCap_EZ_Exome_v2_tiled","nimblegen_SeqCap_EZ_Exome_v2_tiled_extended200","uniovi/ONCOEPIC/V1"]},"probeFile":{"description": "Other probes BED (http://www.ensembl.org/info/website/upload/bed.html#required) file not present in probes catalog","required": false ,"title": "Probe File","type":"string","format":"dreampipe-file"},"mapQual":{"description": "Minimum mapping quality (http://samtools.sourceforge.net/samtools.shtml)","required": true ,"title": "Map Qual","default": 20,"type":"integer"},"baseQual":{"description": "Minimum base quality (http://samtools.sourceforge.net/samtools.shtml)","required": true ,"title": "Base Qual","default": 10,"type":"integer"},"callabilityStatus":{"description": "Callability status of each probe region to be included in the output callabilityBed. Common used values are: All, Callable or LowCoverage","required": true ,"title": "Callability Status","default": "LowCoverage","type":"string","enum":["All","Callable","Uncallable","Strand","LowCoverage","Out"]},"subsamplePercent":{"description": "Percent to extract a random subsample of reads from the BAM (http://samtools.github.io/hts-specs/SAMv1.pdf) file","required": true ,"title": "Subsample Percent","default": 0.01,"type":"number"},"caller":{"description": "Variants caller. Choose between VarScan or DGCaller (DREAMgenics variant caller implementation)","required": false ,"title": "Caller","default": "VarScan","type":"string","enum":["DGCaller","VarScan"]},"BAQcomputation":{"description": "Base Alignment Quality computation (BAQ, is the Phred(http://en.wikipedia.org/wiki/Phred_quality_score)-scaled probability of a read base being misaligned) mode for mpileup command in Samtools (http://samtools.sourceforge.net/samtools.shtml). Set it to [-B, -E or -N]. Which means BAQ computation to be disable, extended or normal respectively","required": true ,"title": "BA Qcomputation","default": "-E","type":"string"},"downgrading":{"description": "Coefficient for downgrading mapping quality of reads containing excessive mismatches (http://samtools.sourceforge.net/samtools.shtml)","required": true ,"title": "Downgrading","default": 50,"type":"integer"},"maxCoverage":{"description": "Max per-BAM depth to avoid excessive memory usage","required": true ,"title": "Max Coverage","default": 100000,"type":"integer"},"minCoverage":{"description": "Minimum read depth at a position to make call variants(http://samtools.sourceforge.net/samtools.shtml)","required": true ,"title": "Min Coverage","default": 6,"type":"integer"},"minAltCount":{"description": "Minimum supporting reads at a position to call variants (http://samtools.sourceforge.net/samtools.shtml) and variant context stats annotation","required": true ,"title": "Min Alt Count","default": 3,"type":"integer"},"minAvgQual":{"description": "Minimum base quality at a position to count a read (http://samtools.sourceforge.net/samtools.shtml)","required": true ,"title": "Min Avg Qual","default": 15,"type":"integer"},"minFreqHomoz":{"description": "Minimum frequency to call homozygote (http://samtools.sourceforge.net/samtools.shtml)","required": true ,"title": "Min Freq Homoz","default": 0.75,"type":"number"},"callerPValue":{"description": "P-value threshold for calling variants (http://samtools.sourceforge.net/samtools.shtml)","required": true ,"title": "Caller P Value","default": 0.05,"type":"number"},"adjVarFreq":{"description": "VarScan adj-var-freq param (Trio calling mode)","required": true ,"title": "Adj Var Freq","default": 0.1,"type":"number"},"adjPValue":{"description": "VarScan adj-p-value param (Trio calling mode)","required": true ,"title": "Adj P Value","default": 0.05,"type":"number"},"callerMode":{"description": "Caller mode: normal or trio","required": false ,"title": "Caller Mode","default": "Normal","type":"string","enum":["Normal","Trio"]},"dgCallerGenomicDatabases":{"description": "Genomic databases used to find matches by DGCaller","required": false ,"title": "Dg Caller Genomic Databases","type":"array","items":{"type":"string"},"enum":["1000genomes/phase3-1","1000genomes/phase3-2","bic/hg19","cll/20140217-3","cosmic/68-1","cosmic/71-1","cosmic/71-2","dbnsfp/2.8-2","dbnsfp/2.9-1","dbnsfp/3.0-1","dbscSNV/20150107-1","dbsnp/all/138-1","dbsnp/all/141-1","dbsnp/all/142-1","dbsnp/all/142-2","dbsnp/all/144-1","dbsnp/clinvar/20140303-1","dbsnp/clinvar/20150115-1","dbsnp/clinvar/20150305-1","dbsnp/clinvar/20150305-2","dbsnp/clinvar/20150629-1","dbsnp/clinvar/20150901-1","dbsnp/clinvar/20150929-1","dbsnp/common/137-1","dbsnp/oncogenicsV4/20150525-1","dgva/20140603-1","dlbcl-shm/20121012-3","esp/6500-1","esp/6500-2","esp/6500-3","exac/0.3-1","exac/0.3-2","hgmd/2014.1-2","hgmd/2014.1-3","hgmd/2015-2","hgmd/2015.1-3","icgc/17-1","icgc/18-1","icgc/19-1","pfam/20140516-1","repeat-masker/20140516-1","telcent/hg19-1","telcent/saccharomyces-cerevisiae/R64-1-1"]},"minCoverageTumor":{"description": "Min coverage for tumour in somatic mode (DGCaller)","required": true ,"title": "Min Coverage Tumor","default": 6,"type":"integer"},"minVariantMedianMQ":{"description": "Min variant median MQ (DGCaller)","required": true ,"title": "Min Variant Median MQ","default": 30,"type":"number"},"minVariantMedianBq":{"description": "Min variant median BQ (DGCaller)","required": true ,"title": "Min Variant Median Bq","default": 20,"type":"number"},"hpSeekExtension":{"description": "Maximun homopolymer base threshold for variant context stats annotation","required": true ,"title": "Hp Seek Extension","default": 8,"type":"integer"},"maxMismatchRatio":{"description": "Max mismatch ratio (DGCaller)","required": true ,"title": "Max Mismatch Ratio","default": 0.035,"type":"number"},"minMarginalFreq":{"description": "Min variant marginal frequency (DGCaller)","required": true ,"title": "Min Marginal Freq","default": 0.4,"type":"number"},"maxClipRatio":{"description": "Maximun clipping ratio per read for variant context stats annotation","required": true ,"title": "Max Clip Ratio","default": 0.5,"type":"number"},"strandPValue":{"description": "Maximun p-value for strand bias for variant context stats annotation","required": true ,"title": "Strand P Value","default": 0.000001,"type":"number"},"maxIndelDisagree":{"description": "Maximun INDEL (http://en.wikipedia.org/wiki/Indel) disagreement ratio at a position for variant context stats annotation","required": true ,"title": "Max Indel Disagree","default": 0.15,"type":"number"},"maxLimitCloseness":{"description": "Variants distance threshold to end of the reads","required": true ,"title": "Max Limit Closeness","default": 3,"type":"number"},"maxWindowContaminationRatio":{"description": "Max window contamination threshold","required": true ,"title": "Max Window Contamination Ratio","default": 0.2,"type":"number"},"pollutionWindowSize":{"description": "Pollution window size (DGCaller)","required": true ,"title": "Pollution Window Size","default": 9,"type":"integer"},"variantPvalue":{"description": "Variant p-value based on Fisher Exact Test","required": true ,"title": "Variant Pvalue","default": 0.05,"type":"number"},"tandemRepeatThreshold":{"description": "Maximun tandem repeat cluster threshold in variant context stats annotation","required": true ,"title": "Tandem Repeat Threshold","default": 8,"type":"integer"},"doProbeDistanceFiltering":{"description": "If checked, do probe distance variant filtering in annotation step ","required": true ,"title": "Do Probe Distance Filtering","default": false,"type":"boolean"},"probeDistanceFilteringValue":{"description": "If flag checked determines the maximum probe distance to keep a variant in annotation step","required": true ,"title": "Probe Distance Filtering Value","default": 50,"type":"integer"},"minClippedBases":{"description": "Minimum number of clipped bases to be keeped","required": true ,"title": "Min Clipped Bases","default": 20,"type":"integer"},"minSdInsertSize":{"description": "Minimun standard deviation","required": true ,"title": "Min Sd Insert Size","default": 10,"type":"number"},"rearrangementsMaxMismatch":{"description": "Max mismatch ratio in rearrangements","required": true ,"title": "Rearrangements Max Mismatch","default": 0.03,"type":"number"},"minSliding":{"description": "Min read sliding to compute internal read duplicates in rearrangements","required": true ,"title": "Min Sliding","default": 10,"type":"integer"},"minSupportingReads":{"description": "Min supporting reads in rearrangements","required": true ,"title": "Min Supporting Reads","default": 8,"type":"integer"},"rearrangementsCallerCoverage":{"description": "Minimum "read coverage" to call rearrangements candidates","required": true ,"title": "Rearrangements Caller Coverage","default": 10,"type":"integer"},"overlappingExtraBases":{"description": "Number of bases around each region to consider overlapping against probe","required": true ,"title": "Overlapping Extra Bases","default": 50,"type":"integer"},"doParallelAnnotation":{"description": "commons.annotation.AnnotationAndIndexationWorkFlow.doParallelAnnotation","required": true ,"title": "Do Parallel Annotation","default": true,"type":"boolean"},"refSeqVersion":{"description": "commons.annotation.AnnotationTask.refSeqVersion","required": false ,"title": "Ref Seq Version","type":"string","enum":["20150611","hg18/20150515","hg19/20150611","hg38/20150511"]},"overlappingRatio":{"description": "Region overlapping ratio for matching used in annotation by overlapping","required": true ,"title": "Overlapping Ratio","default": 0,"type":"number"},"ensemblTranscriptsFile":{"description": "File containing Ensembl transcripts ids. Functional annotation will be applied only over these transcripts","required": false ,"title": "Ensembl Transcripts File","type":"string","format":"dreampipe-file"},"refSeqTranscriptsFile":{"description": "File containing RefSeq transcripts ids. Functional annotation will be applied only over these transcripts","required": false ,"title": "Ref Seq Transcripts File","type":"string","format":"dreampipe-file"},"onlyRelevantTranscripts":{"description": "Limit annotations to only relevant transcripts (CCDS transcripts or longest transcript with biotype miRNA, protein_coding, lincRNA, snRNA, snoRNA, Mt_rRNA, misc_RNA, rRNA, IG_C_gene,IG_D_gene, IG_J_gene, IG_V_gene, TR_C_gene, TR_D_gene, TR_J_gene, TR_V_gene)","required": true ,"title": "Only Relevant Transcripts","default": false,"type":"boolean"},"minQualityRatio":{"description": "Minimun ratio of high quality reads for variant context stats annotation","required": true ,"title": "Min Quality Ratio","default": 0.2,"type":"number"},"maxMismatch":{"description": "Maximun mismatches per read in variant context stats annotation","required": true ,"title": "Max Mismatch","default": 2,"type":"integer"},"maxClusters":{"description": "Maximun number of clusters per window in variant context stats annotation","required": true ,"title": "Max Clusters","default": 2,"type":"integer"},"baseQualFault":{"description": "Minimun "base quality" (http://en.wikipedia.org/wiki/Phred_quality_score) at a position for variant context stats annotation","required": true ,"title": "Base Qual Fault","default": 20,"type":"integer"},"geneSeekWindow":{"description": "If a variant does not fall within a gene, the number of bases in front and behind the variant in which the closest gene is sought","required": true ,"title": "Gene Seek Window","default": 10000,"type":"integer"},"minMedianBq":{"description": "Minimun median base quality per read in variant context stats annotation","required": true ,"title": "Min Median Bq","default": 20,"type":"integer"},"minIndelMarginalFreq":{"description": "Minimun INDEL (http://en.wikipedia.org/wiki/Indel) marginal frequency for variant context stats annotation","required": true ,"title": "Min Indel Marginal Freq","default": 0.75,"type":"number"},"minSnvMarginalFreq":{"description": "Minimun SNV (http://en.wikipedia.org/wiki/Single-nucleotide_polymorphism) marginal frequency for variant context stats annotation","required": true ,"title": "Min Snv Marginal Freq","default": 0.4,"type":"number"},"minEndDist":{"description": "Minimun median distance to the ends of the read for variant context stats annotation","required": true ,"title": "Min End Dist","default": 4.9,"type":"number"},"minVarSd":{"description": "Minimun mutation distribution in the reads (measured as standard deviation (sd)) for variant context stats annotation","required": true ,"title": "Min Var Sd","default": 3,"type":"integer"},"maxAltFoldDiff":{"description": "Maximun strand fold difference for strand bias in variant context stats annotation","required": true ,"title": "Max Alt Fold Diff","default": 5,"type":"number"},"doContextStatsFiltering":{"description": "If checked, discards those variantes with values below params: Min Var Freq, Min Alt Count, Min Coverage","required": false ,"title": "Do Context Stats Filtering","default": false,"type":"boolean"},"doFaultSummaryFiltering":{"description": "If checked, discards variants with faultSumary, wich means that some problems were detected during sequencing or alignment steps (Only if VarScan is used as caller)","required": false ,"title": "Do Fault Summary Filtering","default": false,"type":"boolean"}}},"Inputs":{"description": "Pipeline execution inputs","type": "object","properties": {"dataFolder":{"description": "Root folder containing input data for pipeline. It can contains bcl data structure, fastq files or subfolders (one per sample if is multisample pipeline) containnig fasta files","required": false ,"title": "Data Folder","type":"string","format":"dreampipe-file"},"sample":{"description": "Sample: sampleId and 1 to n pairs of FASTQ (http://en.wikipedia.org/wiki/FASTQ_format) files or a BAM (http://samtools.github.io/hts-specs/SAMv1.pdf)","required": true ,"title": "Sample","type":"object","properties":{"sampleId":{"type":"string"},"fastqPairs":{"type":"array","items":{"type":"object","properties":{"file2":{"type":"string","format":"dreampipe-file"},"file1":{"type":"string","format":"dreampipe-file"}}}},"bam":{"type":"string","format":"dreampipe-file"}}},"trimming":{"description": "If flag is marked, trimming (Trimmomatic - http://www.usadellab.org/cms/?page=trimmomatic) for FASTQ (http://en.wikipedia.org/wiki/FASTQ_format) files is done","required": true ,"title": "Trimming","default": true,"type":"boolean"},"removeDuplicates":{"description": "If flag is marked, removing/marking "PCR duplicates" (http://www.cureffi.org/2012/12/11/how-pcr-duplicates-arise-in-next-generation-sequencing/) from BAM (http://samtools.github.io/hts-specs/SAMv1.pdf) is done","required": true ,"title": "Remove Duplicates","default": true,"type":"boolean"},"doStructural":{"description": "If flag is marked structural variants workflow is done","required": true ,"title": "Do Structural","default": false,"type":"boolean"},"bamStats":{"description": "If flag is marked, provides statistics regarding reads and MQ per Read Group. Bed file or probe version with regions of interest can be provided optionally","required": true ,"title": "Bam Stats","default": true,"type":"boolean"},"zeroBased":{"description": "If flag is marked, probes file is considered to be zero based","required": true ,"title": "Zero Based","default": false,"type":"boolean"},"coverage":{"description": "Minimum "read coverage" (http://en.wikipedia.org/wiki/Deep_sequencing) to compute Callability (Measure of a genotypes position confidence based on "mapping quality" (http://genome.sph.umich.edu/wiki/Mapping_Quality_Scores), "base quality" (http://en.wikipedia.org/wiki/Phred_quality_score) and "read coverage" (http://en.wikipedia.org/wiki/Deep_sequencing) thresholds) ","required": true ,"title": "Coverage","default": [10,20,50,100,200,500,1000],"type":"array","items":{"type":"integer"}},"callabilityStatusCoverage":{"description": "Coverage that will be used to create a bed file containing all regions whose callability status matches the desired in paramter callabilityStatus","required": false ,"title": "Callability Status Coverage","default": 20,"type":"integer"}}},"Tags":{"description": "Execution tags. Categorizes the execution", "type":"array","items":{"type":"string"}},"Emails":{"description": "Email addresses to be notified of execution events", "type":"array","items":{"type":"string"}}}}